Ретинобластома - наиболее часто встречающаяся внутриглазная злокачественная опухоль детского возраста (1-45), развивающаяся приблизительно у 1 из 15000 живых новорожденных. Эта злокачественная опухоль развивается скрыто, проявляется лишь безболезненной лейкокорией и приводит к гибели пациента (1,2). При отсутствии лечения летальный исход наступает в течение 1-2 лет. Массивные опухоли, прорастающие окружающие ткани, характеризуются наибольшим риском метастазирования. Во всем мире выживаемость при ретинобластоме коррелирует с уровнем экономического развития: в Африке она составляет приблизительно 30%, 60% - в Азии, 80% - Латинской Америке, и от 95% до 97% в Европе и Северной Америке (1).
а) Генетика. Причиной ретинобластомы является соматическая или зародышевая мутация 13 хромосомы (3-8). При всех двусторонних и семейных формах ретинобластомы имеются зародышевые мутации. При односторонней ретинобластоме зародышевая мутация выявляется в 15% случаев и соматическая мутация - в 85% случаев. У пациентов с зародышевой мутацией может присутствовать видимые клинические проявления синдрома 13q хромосомы; также у них отмечается высокий риск развития пинеа-лобластомы и других злокачественных опухолей.
Пинеалобластома и другие параселлярные опухоли с точки зрения анатомии, эмбриологии и иммунологии идентичны ретинобластоме (9-16). У пациентов с зародышевой мутацией отмечена тенденция к развитию этих злокачественных опухолей, обычно они возникают в течение года после диагностики ретинобластомы и почти всегда в возрасте моложе пяти лет (14). Сочетание двусторонней ретинобластомы и пинеалобластомы получило название трилатеральной ретинобластомы. Но этот термин не совсем точен, поскольку у некоторых пациентов с пинеалобластомой ретинобластома развивается только с одной стороны или не развивается вовсе.
С другой стороны, другие злокачественные опухоли у таких больных могут развиваться в течение всей жизни (17-21), наиболее часто это остеосаркома длинных костей, саркома мягких тканей и меланома кожи. Если пациенты с зародышевой мутацией выживают после второй злокачественной опухоли, у них также сохраняется риск развития третьей, четвертой и пятой опухолей. У детей, получавших системную химиотерапию по поводу ретинобластомы, отмечается снижение риска развития других злокачественных опухолей в отдаленном периоде (21).
б) Клиническая картина. Клинические признаки ретинобластомы варьируют в зависимости от распространенности опухоли (22-45). В большинстве случаев опухоль выявляется у детей в возрасте младше трех лет. Однако эта опухоль может диагностироваться и в более старшем возрасте, у подростков или даже у взрослых (29, 30). В Соединенных Штатах первыми симптомами опухоли чаще всего являются лейкокория (56%), косоглазие (24%) и низкие зрительные функции (8%) (22). В более позднем исследовании другой группы пациентов, проведенном в США и включавшем в себя около 1200 глаз, средний возраст на момент манифестации заболевания составил 15 месяцев, 51% составили пациенты мужского пола, 49% - женского, одностороннее поражение наблюдалось в 53% случаев, в 47% диагностирована двусторонняя опухоль (23). Другие крайние цифры были получены в некоторых областях Африки, где опухоль чаще всего манифестирует буфтальмом (56%) и лейкокорией (32%), при этом отмечается высокий риск двусторонней энуклеации и летального исхода (24).
Клинически ретинобластома возникает в виде мелкого прозрачного новообразования сенсорной сетчатки, которое можно легко не заметить при офтальмоскопии. По мере увеличения опухоли она становится матово-белой, появляются расширенные ретинальные питающая артерия и дренирующая вена, может развиваться вторичная отслойка сетчатки. Среди первых симптомов наиболее известен белый зрачковый рефлекс, получивший название лейкокория. По мере роста опухоли она перестает быть исключительно интраретинальным новообразованием и демонстрирует экзофитный рост, эндофитный рост, или комбинацию этих двух типов роста. Экзофитный рост характеризуется увеличением опухоли кнаружи, в субретинальное пространство, что вызывает отслойку вышележащей сетчатки. Эндофитный рост характеризуется появлением в стекловидном теле отсевов опухолевых клеток, иногда препятствующих визуализации сетчатки. Изредка в ткани ретинобластомы может формироваться внутренняя полость, что указывает на высокую дифференцировку опухоли (36).
Реже наблюдается диффузный инфильтративный рост, характеризуемый развитием плоской или минимально проминирующей опухоли (31); хотя диффузная ретинобластома обычно локализуется в задних отделах сетчатки, изредка она развивается вблизи ora serrata и над цилиарным телом и не прорастает расположенные сзади структуры (32). Ретинобластома становится причиной вторичной глаукомы приблизительно в 17% случаев, обычно вследствие неоваскуляризации радужки и вторичного закрытия угла передней камеры (33). Неоваскуляризация радужки может сопровождаться спонтанными гифемами, которые изредка являются первым симптомом опухоли. Воспаление, вызванное некротической внутриглазной ре-тинобластомой, может симулировать или провоцировать целлюлит глазницы (34). Хотя обычно сохраняются прозрачность и правильное положение хрусталика, в редких случаях развиваются катаракта и подвывих хрусталика. В далекозашедших случаях наблюдается экстрасклераль-ное распространение опухоли, которая выглядит как объемная эрозивная масса.
РЕТИНОБЛАСТОМА: КЛИНИЧЕСКАЯ КАРТИНА
На ранних стадиях ретинобластома представляет собой мелкое прозрачное новообразование сетчатки. Несколько увеличившись, она становится матовой и более заметной, со временем появляются расширенные сосуды - питающая ретинальная артерия и дренирующая вена. Иногда при офтальмоскопии в ткани опухоли визуализируются очаги кальциноза «мелового» белого цвета. Опухоли фовеолярной области вызывают нарушение фиксации взора и развитие косоглазия, как эзотропии, так и экзотропии. Со временем развивается характерный белый зрачковый рефлекс (лейкокория). В большинстве случаев опухоль диагностируется уже после развития лейкокории.
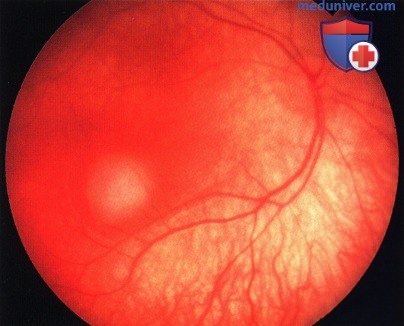
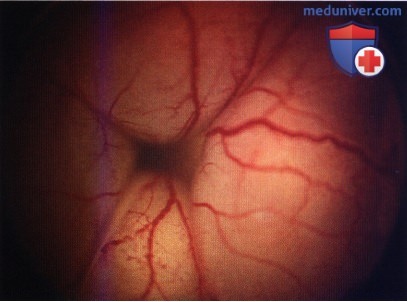
Мелкая ретинобластома снизу от центральной ямки правого глаза.
Несколько более крупная ретинобластома сверху от диска зрительного нерва. Обратите внимание на расширенные сосуды - питающую артерию и дренирующую вену.
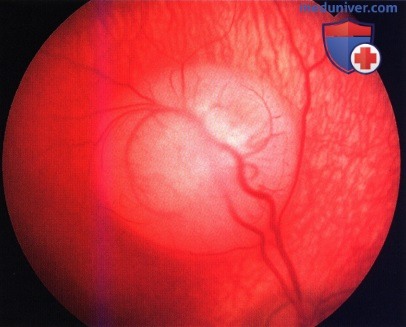
Более крупная ретинобластома в верхней части макулярной зоны. Обратите внимание на расширение сосудов верхней аркады, идущих от диска зрительного нерва, при этом сосуды нижней аркады не расширены.
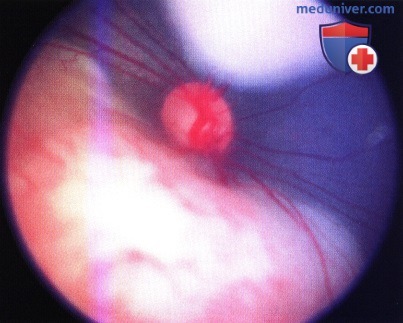
Две ретинобластомы, прилегающие к диску зрительного нерва. Верхняя опухоль диссеминирует в вышележащее стекловидное тело (эндофитный рост), нижняя опухоль все еще лежит в плоскости сетчатки.
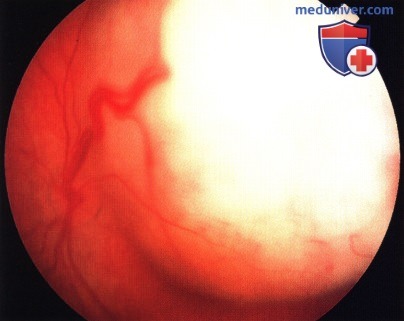
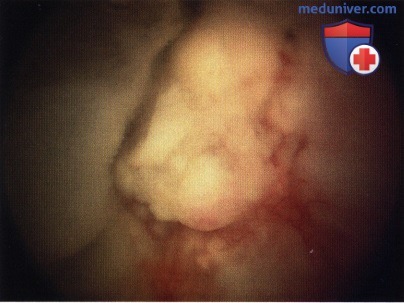
Массивная ретинобластома эндофитного типа.
Массивная ретинобластома экзофитного типа.
в) Спонтанный регресс. Приблизительно в 3% случаев отмечается тенденция к спонтанному регрессу ретинобластомы (37-40). Первоначально была описана спонтанно регрессировавшая ретинобластома, имевшая достаточно типичные клинические признаки. Впоследствии был описан доброкачественный вариант ретинобластомы, названный рети-ноцитомой или ретиномой. Для обозначения сначала увеличивавшейся до определенного размера, а затем самостоятельно регрессировавшей опухоли мы предпочитаем пользоваться термином спонтанно регрессировавшая ретинобластома. Доброкачественный вариант опухоли, увеличивавшейся до определенного размера, а затем стабилизировавшейся, мы называем спонтанно стабилизировавшаяся ретинобластома (1).
г) Международная классификация ретинобластомы. В настоящее время в крупных лечебных центрах, занимающихся лечением таких пациентов используется Международная классификация ретинобластомы (41-43) (табл. 15.1). Нюансы этой классификации и их использование для прогнозирования и оценки перспектив сохранения глаза после проведения хеморедукции и интраартериальной химиотерапии обсуждаются в соответствующих публикациях (44, 45). Классификация сведена в таблицу, которую мы приводим ниже.
Ниже описаны и проиллюстрированы клинические варианты, диагностика, лечение и дифференциальная диагностика ретинобластомы.
д) Список использованной литературы:
1. Kivela Т. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 2009;93:1129-1131.
2. Wong JR, Tucker MA, Kleinerman RA, et al. Retinoblastoma incidence patterns in the US Surveillance, Epidemiology, and End Results program. JAMA Ophthalmol 2014;132:478-483.
3. Sparkes RS, Murphree AL, Lingua RW, et al. Gene for hereditary retinoblastoma assigned to human chromosome 13 by linkage to esterase D. Science 1983;219:971-973.
4. Shields CL, Shields JA, Donoso I,A. Clinical genetics of retinoblastoma. In: Shields (A, ed. Update on Malignant Ocidar Tumors. Boston: Little, International Ophthalmology Clinics, Brown; 1993;33:67-76.
5. Ganguly A, Nichols K, Grant G, et al. Molecular karyotype of sporadic unilateral retinoblastoma tumors. Retina. 2009;29:1002-1012.
6. Ganguly A, Shields CL. Differential gene expression profile of retinoblastoma compared to normal retina. Mol Vis 2010;16:1292-1303.
7. Nichols KE, Walther S, Chao E, et al. Recent advances in retinoblastoma genetic research. Curr Opin Ophthalmol 2010;20:351-355.
8. Chen A, Moran K, Richard-Yutz J, et al. Enhanced sensitivity for detection of lowlevel germline mosaic RBI mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat 2013;35(3):384-391.
9. Donoso LA, Shields JA, Felberg NT, et al. Intracranial malignancy in patients with bilateral retinoblastoma. Retina 1981;1:67-74.
10. Bader JL, Meadows AT, Zimmerman LE, et al. Bilateral retinoblastoma with ectopic intracranial retinoblastoma: trilateral retinoblastoma. Cancer Genet Cytogenet 1982;5: 203-213.
11. De Potter P, Shields CL, Shields JA. Clinical variations of trilateral retinoblastoma. A report of 13 cases. J Pediatr Ophthalmol Strabismus 1994;31:26-31.
12. Marcus DM, Brooks SE, Leff G, et al. Trilateral retinoblastoma: insights into histogenesis and management. Surv Ophthalmol 1998;43:59-70.
13. Pesin SR, Shields JA. Seven cases of trilateral retinoblastoma. Am J Ophthalmol 1989;107:121-126.
14. Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma .J Clin Oncol 1999;17: 1829-1837.
15. Singh AD, Shields CL, Shields JA. New insights into trilateral retinoblastoma. Cancer 1999;86:3-5.
16. Ramasubramanian A, Kytasty C, Meadows AT, et al. Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 2013; 156(4):825—829.
17. Abramson DH, Ellsworth RM, Zimmerman LE. Nonocular cancer in retinoblastoma survivors. Trans Am Acad Ophthalmol 1976;81:454-456.
18. Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in patients with bilateral retinoblastoma. Ophthalmology 1988;95:1583-1587.
19. Turaka K, Shields CL, Leahey A, et al. Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 2012;59:121-125.
20. Abramson DH, Melson MR, Dunkel IJ, et al. Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmology 2001;108:1868-1876.
21. Moll AC, Imhof SM, Bouter LM, et al. Second primary tumors in patients with hereditary retinoblastoma: a register-based follow-up study, 1945— 1994. lnt J Cancer 1996;67:515-519.
22. Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr 1998;132:505-508.
23. Epstein I, Shields CL, Shields JA. Trends in the management of retinoblastoma; Evaluation of 1,196 consecutive eyes during 1974-2001. I Ped Ophthalmol Strabismus 2003;40:196-203.
24. Boubacar Т, Fatou S, Fousseyni Т, et al. A 30-month prospective study on the treatment of retinoblastoma in the Gabriel Toure Teaching Hospital, Bamako, Mali. Br J Ophthalmol 2010;94:467-469.
25. Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol 2006;17:228-234.
26. Shields CL, Fulco EM, Arias JD, et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye (Lond) 2013;27(2): 253-264.
27. Shields CL, Shields JA. Pearls in the management of children with retinoblastoma. Saudi J Ophthalmol 2009;23:43-50.
28. Shields CL, Schwendeman R, Lally SE, et al. Targeted retinoblastoma management. When to use intravenous, intra-arterial, subTenons, and intravitreal chemotherapy. Curr Opin 2014;25(5):374-385.
29. Shields CL, Shields JA, Shah P. Retinoblastoma in older children. Ophthalmology 1991;98:395-399.
30. Kaliki S, Shields CL, Gupta A, et al. Newly-diagnosed active retinoblastoma in adults. A study of 8 cases. Retina 2015; in press.
31. Shields CL, Ghassemi F, Tuncer S, et al. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthalmology 2008;115:2253-2258.
32. Grossniklaus HE, Dhaliwal RS, Martin DF. Diffuse anterior retinoblastoma. Retina 1998;18:238-241.
33. Shields CL, Shields JA, Shields MB, et al. Prevalence and mechanisms of secondary intraocular pressure elevation in eyes with intraocular tumors. Ophthalmology 1987;94:839-846.
34. Shields JA, Shields CL, Suvarnamani C, et al. Retinoblastoma manifesting as orbital cellulitis. Tenth Annual David and Mary Seslen Endowment Lecture. Am J Ophthalmol 1991; 112:442-449.
35. Shields CL, Piccone MR, Shields JA, et al. Mushroom-shaped choroidal recurrence of retinoblastoma 25 years after therapy. Arch Ophthalmol 2002;120:844-846.
36. Palamar M, Pirondini C, Shields CL, et al. Cavitary retinoblastoma. Ultrasonography and fluorescein angiographic findings in 3 cases. Arch Ophthalmol 2008; 126(11): 1598-1600.
37. Gallie BL, Ellsworth RM, Abramson DH, et al. Retinoma: spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer 1982;45: 513-521.
38. Margo C, Hidayat A, Kopelman J, et al. Retinocytoma. A benign variant of retinoblastoma. Arch Ophthalmol 1983; 101;1519-1531.
39. Eagle RC, Shields JA, Donoso LA, et al. Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology 1989;96:1389-1395.
40. Singh AD, Santos MC, Shields CL, et al. Observations on 17 patients with retinocytoma. Arch Ophthalmol 2000; 118:199-205.
41. Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol 2006;113:2080-2086.
42. Shields CL. The International Classification of Retinoblastoma is practical and predictable. In: Rapuano C, ed, Yearbook of Ophthalmology. St Louis, MO: Mosby; 2008; 227-230.
43. Chantada GL, Sampor C, Bosaleh A, et al. Comparison of staging systems for extraocular retinoblastoma: analysis of 533 patients. JAMA Ophthalmol 2013; 131 (9): 1127-1134.
44. Shields CL, Au A, Czyz C, et al. The International Classification of Retinoblastoma (ICRB) predicts chemoreduction success. Ophthalmology 2006;113:2276-2280.
45. Shields CL, Manjandavida FP, Pieretti G, et al. Intra-arterial chemotherapy for retinoblastoma in 70 eyes: Outcomes based on the International Classification of Retinoblastoma. Ophthalmology 2014;121(7):1453-1460.