Коагулопатии - кратко с точки зрения внутренних болезней
I. Нарушения первичного гемостаза
Первоначальное образование тромбоцитарного сгустка (см. рис. ниже, А, также известное как первичный гемостаз) может нарушаться при тромбоцитопении, болезни Виллебранда, а также при тромбоцитопатиях и заболеваниях, поражающих сосудистую стенку.
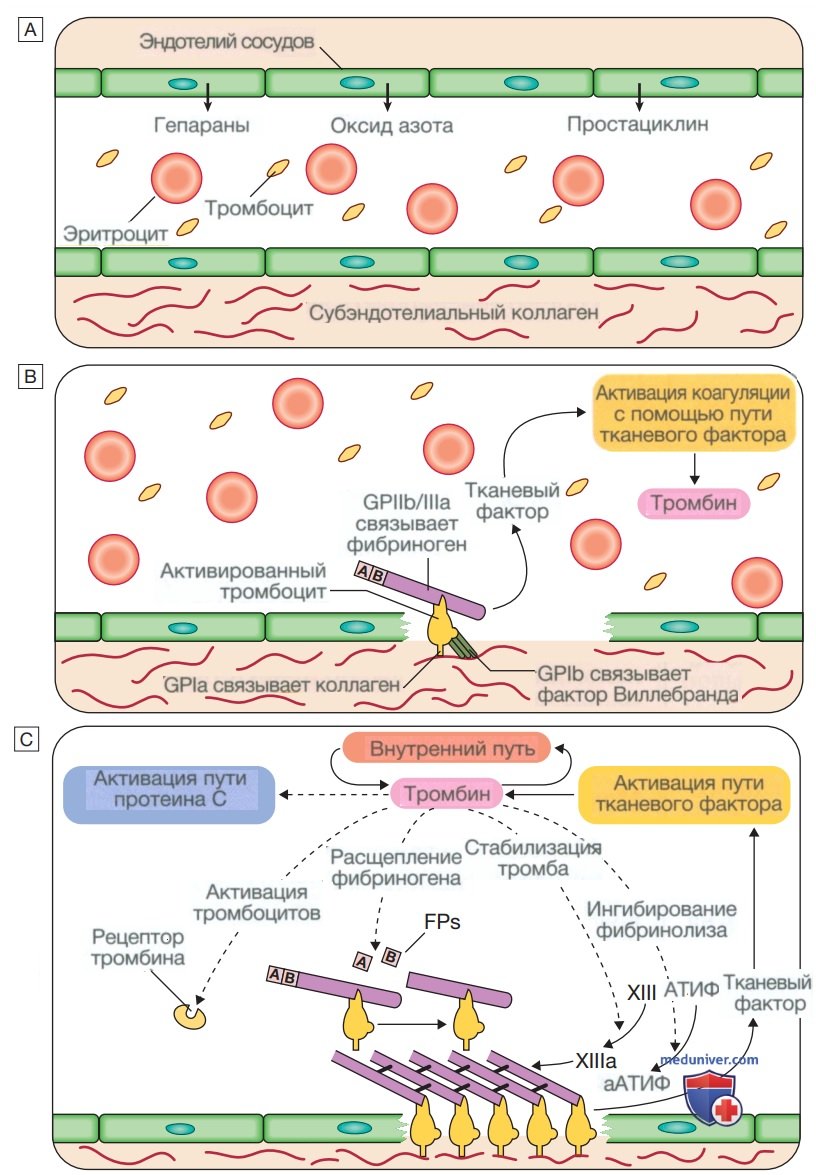
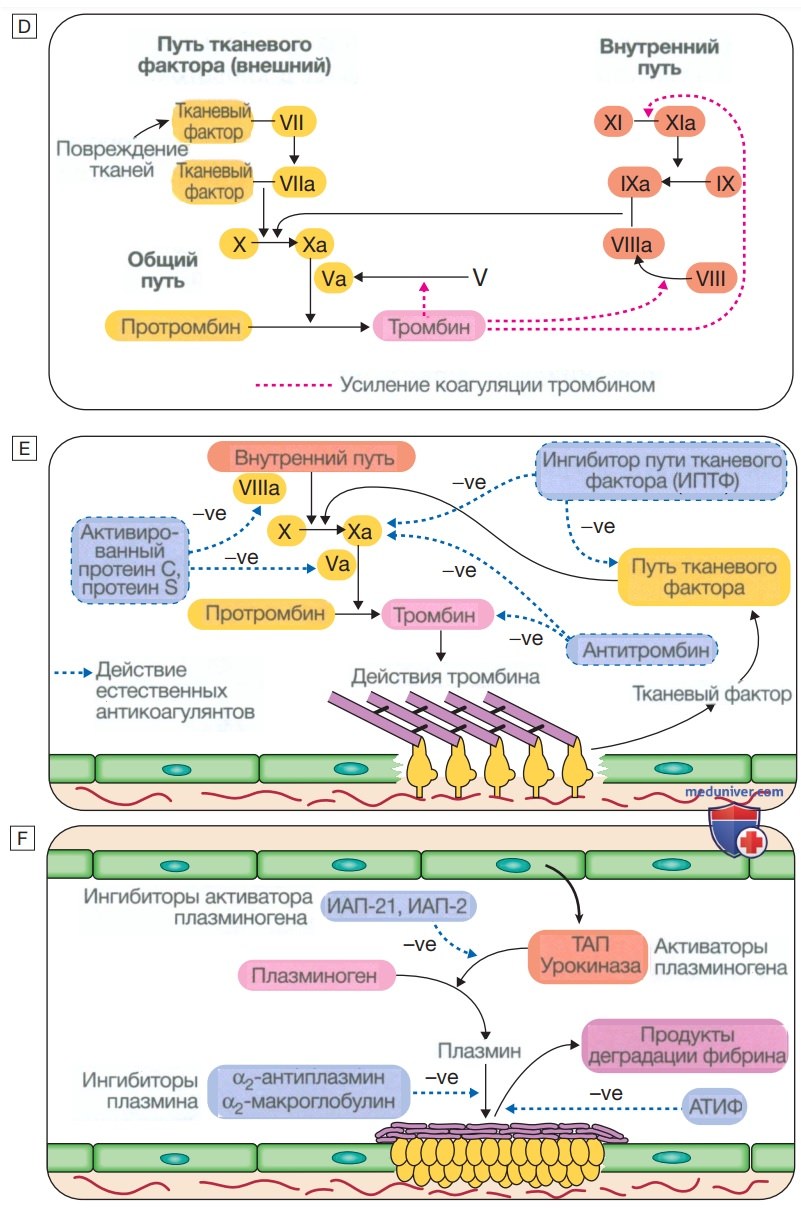
Стадии гемостаза в норме. А — стадия 1. Условия перед травмой не мешают кровотоку. Эндотелий сосудов вырабатывает вещества (в том числе оксид азота, простациклин и гепариноподобные вещества) для предотвращения адгезии тромбоцитов и лейкоцитов к стенке сосуда. Тромбоциты и факторы свертывания крови циркулируют в неактивном состоянии. В — стадия 2. Ранний гемостатический ответ: тромбоциты адгезируются; активируется коагуляция. В месте повреждения сосуда эндотелий разрушается, обнажая субэндотелиальный коллаген. Выделяются небольшие количества тканевого фактора (ТФ). Тромбоциты связываются с коллагеном с помощью специфического рецептора гликопротеина la (GPIa), что вызывает изменение формы тромбоцитов и их адгезию к области повреждения за счет связывания других рецепторов (GPIb и GPIIb/llla) с фактором Виллебранда и фибриногеном соответственно. Коагуляция активируется тканевым фактором (внешний путь) с образованием небольшого количества тромбина. С и D — стадия 3. Формирование фибринового сгустка: тромбоциты активируются и агрегируют; образование фибрина поддерживается мембраной тромбоцитов: образуется стабильный фибриновый сгусток. Адгезирующиеся тромбоциты активируются многими путями, включая связывание аденозиндифосфата, коллагена, тромбина и адреналина (эпинефрина) с поверхностными рецепторами. Циклооксигеназный путь превращает арахидоновую кислоту из мембраны тромбоцитов в тромбоксан А2, который вызывает агрегацию тромбоцитов. Активация тромбоцитов приводит к высвобождению содержимого их гранул, что еще больше усиливает коагуляцию (см. рис. ниже). Тромбин играет основную роль в контроле коагуляции: небольшое количество, образующееся через путь тканевого фактора, значительно усиливает его собственный синтез; «внутренний» путь становится активным с образованием большого количества тромбина. Тромбин непосредственно вызывает образование тромба за счет отщепления фибринопептидов (ФП) от фибриногена с образованием фибрина. Мономеры фибрина «сшиты» фактором XIII, который также активируется тромбином.
(Окончание) Сыграв ключевую роль в образовании и стабилизации тромба, тромбин начинает регулировать образование тромба двумя основными путями: (а) активация пути протеина С (PC) (естественный антикоагулянт), который уменьшает дальнейшую коагуляцию; (b) активация активируемого тромбином ингибитора фибринолиза (АТИФ), который ингибирует фибринолиз (см. Е и F). Д — стадия 4. Ограничение образования тромба: естественные антикоагулянты прекращают активацию факторов свертывания крови. После достижения гемостаза дальнейшее увеличение тромба ограничивается антикоагулянтами. Антитромбин — это ингибитор сериновой протеазы, синтезируемый в печени, который разрушает активированные факторы, такие как XIa, Ха и тромбин (IIа). Его основная активность против тромбина и Ха усиливается гепарином и фондапаринуксом натрия, что объясняет их антикоагулянтный эффект. Ингибитор пути тканевого фактора (ИПТФ) связывается с факторами Vila и Ха и инактивирует их. Активация протеина С происходит после связывания тромбина с мембраносвязанным тромбомодулином; активированный протеин С (аРС) связывается со своим кофактором, протеином S (PS), и расщепляет Va и VIIIa. Протеин С и протеин S зависят от витамина К, и их запасы истощаются кумариновыми антикоагулянтами, такими как варфарин. Е — стадия 5. Фибринолиз: плазмин разрушает фибрин, чтобы обеспечить реканализацию сосуда и восстановление тканей. Нерастворимый тромб необходимо разрушить для реканализации сосуда. Плазмин, основной фибринолитический фермент, образуется при активации плазминогена, например тканевым активатором плазминогена (ТАП) или урокиназой в тромбе. Плазмин гидролизует фибриновый тромб с образованием продуктов деградации фибрина, в том числе D-димера. Этот процесс строго регулируется; активаторы плазминогена контролируются ингибитором, называемым ингибитором активатора плазминогена (ИАП), активность плазмина ингибируется α2-антиплазмином и α2-макроглобулином, а фибринолиз дополнительно ингибируется активируемым тромбином ингибитора фибринолиза
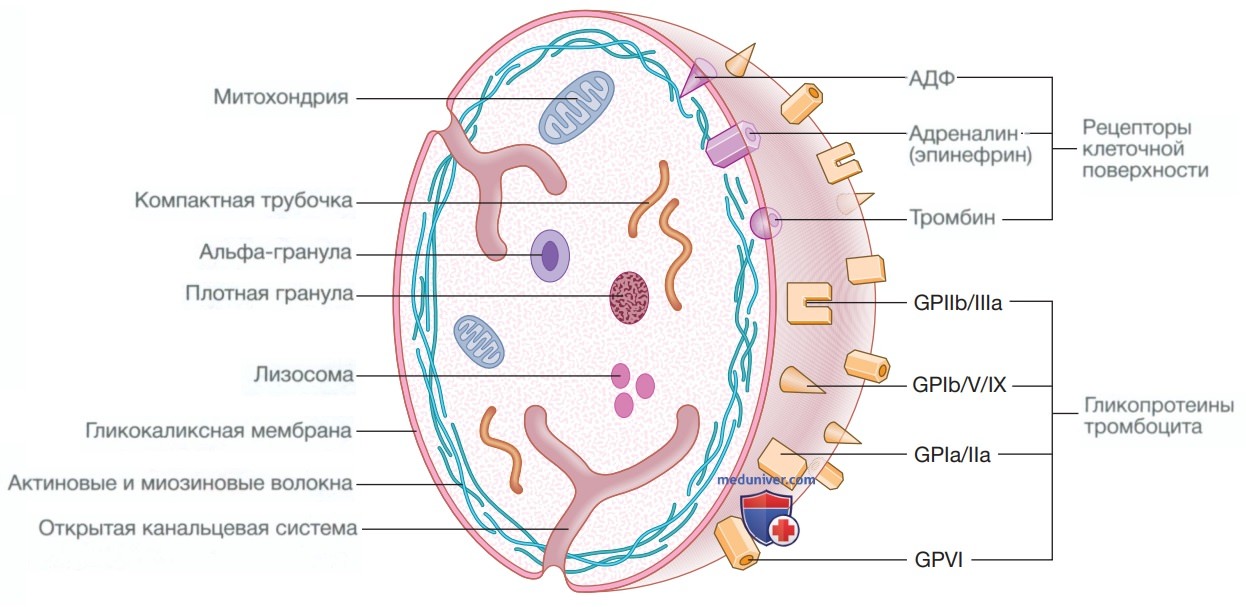
Структура тромбоцита в норме. Поверхность тромбоцита покрыта гликопротеинами, которые связываются с основными структурами, включая фибриноген, коллаген и фактор Виллебранда, а также рецепторами клеточной поверхности для тромбина, аденозиндифосфата и адреналина (эпинефрина). Через внутренние пути передачи сигналов активация тромбоцита вызывает дегрануляцию альфа-гранул и плотных гранул, что в конечном итоге приводит к агрегации тромбоцитов. Блокада этих путей такими препаратами, как ацетилсалициловая кислота (Аспирин), клопидогрел, тикагрелор, тирофибан и абциксимаб, составляет основу антиагрегантной терапии. ГП — гликопротеин
а) Патология сосудистой стенки. Патология сосудистой стенки может быть:
• врожденной (например, наследственная геморрагическая телеангиэктазия);
• приобретенной (например, при васкулите или цинге).
1. Наследственная геморрагическая телеангиэктазия. Наследственная геморрагическая телеангиэктазия (син.: болезнь Рандю—Ослера) наследуется по аутосомно-доминантному типу и вызывается мутациями в генах, кодирующих эндоглин и рецептор активин-подобной киназы, которые являются рецепторами эндотелиальных клеток для трансформирующего фактора роста-β, являющегося мощным ангиогенным цитокином.
Телеангиэктазии и небольшие аневризмы обнаруживаются на кончиках пальцев, лице и языке, а также в носовых ходах, легких и желудочно-кишечном тракте. У значительной части этих пациентов развиваются более крупные легочные артериовенозные мальформации, которые вызывают артериальную гипоксемию из-за сброса крови справа налево. Они предрасполагают к развитию парадоксальных эмболий, приводящих к инсульту или абсцессу головного мозга.
Все пациенты с наследственной геморрагической телеангиэктазией должны быть обследованы для исключения легочных артериовенозных мальформаций; при их обнаружении следует рассмотреть возможность их абляции путем чрескожной эмболизации.
У пациентов отмечаются рецидивирующие кровотечения (в особенности носовые) или дефицит железа вследствие латентных желудочно-кишечных кровотечений. Лечение может быть затруднено из-за множественных источников кровотечения, но постоянная терапия препаратами железа обычно позволяет костному мозгу компенсировать кровопотерю. Местное прижигание или лазеротерапия могут предотвратить кровотечения некоторых локализаций. Были опробованы различные виды медикаментозной терапии, но ни один из них не был признан универсально эффективным.
2. Синдром Элерса-Данло. Сосудистый синдром Элерса—Данло (тип 4) представляет собой редкое аутосомно-доминантное заболевание (1 на 100 000 человек), вызванное дефектом коллагена 3-го типа, который приводит к хрупкости кровеносных сосудов и мембран органов, что вызывает кровотечения и разрывы органов. Классическая гипермобильность суставов часто не выражена при данной форме заболевания, но кожные изменения и внешний вид лица довольно типичны. Данный синдром следует включать в дифференциальный диагноз при наличии в анамнезе кровотечений на фоне нормальных результатов лабораторных исследований.
3. Цинга. Дефицит витамина С нарушает нормальный синтез коллагена и приводит к развитию коагулопатии, характеризующейся перифолликулярными, петехиальными, субпериостальными кровоизлияниями и синяками. В диагностике помогает изучение рациона питания.
б) Нарушения функции тромбоцитов. Кровотечение может быть следствием развития тромбоцитопении (см. табл. 14), а также врожденных или приобретенных нарушений функции тромбоцитов. Наиболее часто приобретенные нарушения являются ятрогенными в результате применения ацетилсалициловой кислоты (Аспирина*), клопидогрела, тикагрелора, дипиридамола и ингибиторов гликопротеина IIb/IIIa, назначаемых для профилактики артериальных тромбозов (см. табл. 26). Наследственные тромбоцитопатии возникают относительно редко.
Врожденные аномалии могут быть связаны с дефицитом мембранных гликопротеинов (например, тромбастения Гланцмана (IIb/IIIa) или синдром Бернара—Сулье (Ib)) или с наличием дефектных гранул тромбоцитов (например, дефицит плотных (дельта) гранул, см. рис. выше), что приводит к нарушению развития пула тромбоцитов.
Врожденные макротромбоцитопатии, обусловленные мутациями в гене тяжелой цепи миозина MYH-9, характеризуются большими тромбоцитами, включениями в нейтрофилах (тельца Деле; известные также как тельца Князькова-Деле) и рядом других проявлений, включая нейросенсорную тугоухость и патологию почек. Другие наследственные тромбоцитопатии также требуют внимания, так как они могут сопровождаться клинической картиной, а некоторые связаны со склонностью к развитию недостаточности или дисплазии костного мозга (например, RUNX-1-ассоциированная тромбоцитопения).
За исключением тромбастении Гланцмана эти нарушения являются незначительными, кровотечения обычно возникают после травмы или операции, но редко бывают спонтанными. Тромбастения Гланцмана — аутосомно-рецессивное заболевание, характеризующееся различными и зачастую тяжелыми нарушениями свертывания крови. Эти заболевания обычно лечат с помощью локального механического воздействия, но также могут быть эффективны антифибринолитичекие препараты, такие как транексамовая кислота, а при тяжелом кровотечении — переливание тромбоцитарной массы. Рекомбинантный фактор Vila одобрен к применению для лечения резистентного кровотечения при тромбастении Гланцмана.
в) Тромбоцитопения. Тромбоцитопения возникает при различных заболеваниях, представленных в табл. 14, многие из которых обсуждаются в отдельных статьях на сайте - просим пользоваться формой поиска по сайту выше.
1. Идиопатическая тромбоцитопеническая пурпура. Идиопатическая тромбоцитопеническая пурпура — иммуноопосредованное заболевание, при котором аутоантитела вырабатываются чаще всего против гликопротеина тромбоцитов IIb/IIIa с последующей сенсибилизацией тромбоцитов и преждевременного удаления их из кровотока клетками ретикулоэндотелиальной системы. Это полиморфное заболевание; в некоторых случаях оно протекает изолированно, в то время как в других — связана с нарушением иммунитета при таких состояниях, как заболевания соединительной ткани, ВИЧ-инфекция, В-клеточные злокачественные новообразования, беременность и на фоне приема ряда ЛС.
Тем не менее клиническая картина и патогенез аналогичны вне зависимости от причины идиопатической тромбоцитопенической пурпуры.
- Клинические проявления, лабораторные и инструментальные исследования. Клиническая картина зависит от степени выраженности тромбоцитопении. Спонтанное кровотечение обычно происходит только тогда, когда число тромбоцитов становится ниже 20 х 109/л. При более высоких показателях пациент может жаловаться на небольшие синяки, иногда на носовые кровотечения или меноррагии. В большинстве случаев тромбоцитопения с уровнем тромбоцитов более 50 х 109/л обнаруживается случайно.
Среди взрослых идиопатической тромбоцитопенической пурпурой чаще болеют женщины, и заболевание может развиваться постепенно. В отличие от идиопатической тромбоцитопенической пурпуры у детей, предшествующая вирусная инфекция в анамнезе выявляется редко. Симптомы или признаки поражения соединительной ткани могут быть заметными на момент постановки диагноза или появиться через несколько лет. Пациентам в возрасте старше 65 лет следует проводить исследование костного мозга для поиска сопутствующего В-клеточного злокачественного новообразования, а также соответствующие исследования на аутоантитела при подозрении на диффузное заболевание соединительной ткани.
Тестирование на ВИЧ-инфекцию следует включать в план обследования, поскольку положительный тест на ВИЧ может оказать существенное влияние на проводимую в дальнейшем терапию. Мазок периферической крови не выявляет патологии, за исключением значительного снижения числа тромбоцитов, в то время как в костном мозге обнаруживается явное увеличение числа мегакариоцитов.
- Лечение. Многим пациентам со стабильной компенсированной идиопатической тромбоцитопенической пурпурой и числом тромбоцитов более 30 х 109/л лечение для повышения уровня тромбоцитов не требуется за исключением случаев повышенного риска кровотечения, таких как хирургическое вмешательство и биопсия.
Терапия первой линии для пациентов со спонтанным кровотечением включает высокие дозы глюкокортикостероидов, преднизолона (1 мг/кг в сутки) либо дексаметазона (40 мг/сут в течение 4 дней) для подавления образования антител и ингибирования фагоцитоза сенсибилизированных тромбоцитов ретикулоэндотелиальными клетками. Введение внутривенного иммуноглобулина может увеличить количество тромбоцитов за счет блокирования рецепторов антител на ретикулоэндотелиальных клетках, а при тяжелой гемостатической недостаточности его можно назначать в комбинации с глюкокортикостероидами, особенно при симптомах значительного кровотечения из слизистых оболочек или при медленном ответе на монотерапию глюкокортикостероидами.
При персистирующем или жизнеугрожающем кровотечении в дополнение к другим методам лечения следует переливать тромбоцитарную массу.
Заболевание может стать хроническим с чередованием ремиссий и рецидивов. Рецидивы следует лечить с помощью повторного введения глюкокортикостероидов. У пациентов с двумя рецидивами или первично рефрактерных к лечению рассматривается терапия второй линии. К ней относят агонисты рецепторов тромбопоэтина, элтромбопаг и ромиплостим, спленэктомию и иммуносупрессивную терапию.
При планировании спленэктомии необходимо соблюдать меры предосторожности, перечисленные в табл. 37. Спленэктомия вызывает полную ремиссию примерно у 70% пациентов и улучшение еще у 20—25% при благоприятном течении заболевания. Агонисты рецепторов тромбопоэтина вызывают ответ примерно в 75% случаев обычно в течение 10—14 дней. Низкие дозы глюкокортикостероидов и иммунодепрессантов (таких как ритуксимаб, циклоспорин, микофенолата мофетил и такролимус) также могут вызывать ремиссию.
Алгоритм назначения терапии не до конца ясен, несмотря на то, что агонисты рецепторов тромбопоэтина одобрены к применению по данному показанию, в то время как иммунодепрессанты — нет.
II. Нарушения свертываемости крови
Процесс свертывания крови в норме объясняется на рис. выше (прямо в начале данной статьи). Дефицит факторов свертывания может быть врожденным и приобретенным и иметь отношения к одному или нескольким факторам (табл. 62). Наследственные нарушения почти всегда связаны со снижением синтеза в результате мутации в гене, кодирующем ключевой белок при коагуляции. Болезнь Виллебранда является наиболее распространенной наследственной коагулопатией. Гемофилия А и В — наиболее частые заболевания, сопровождающиеся дефицитом одного фактора свертывания крови, но описаны наследственные дефициты и всех остальных факторов свертывания.
Приобретенные нарушения могут быть вызваны недостаточным образованием (например, при печеночной недостаточности), повышенным потреблением (например, при ДВС-синдроме) или ингибированием функции факторов свертывания крови (например, на фоне терапии гепарином или вследствие воздействия иммунных ингибиторов свертывания крови, как при приобретенной гемофилии А).
а) Гемофилия А. Дефицит фактора VIII, приводящий к гемофилии А, встречается у 1 на 10 000 человек. Это наиболее распространенный врожденный дефицит фактора свертывания крови. Фактор VIII синтезируется преимущественно печенью и эндотелиальными клетками и имеет период полураспада около 12 ч. Он защищен от протеолиза в кровотоке за счет связывания с vWF.
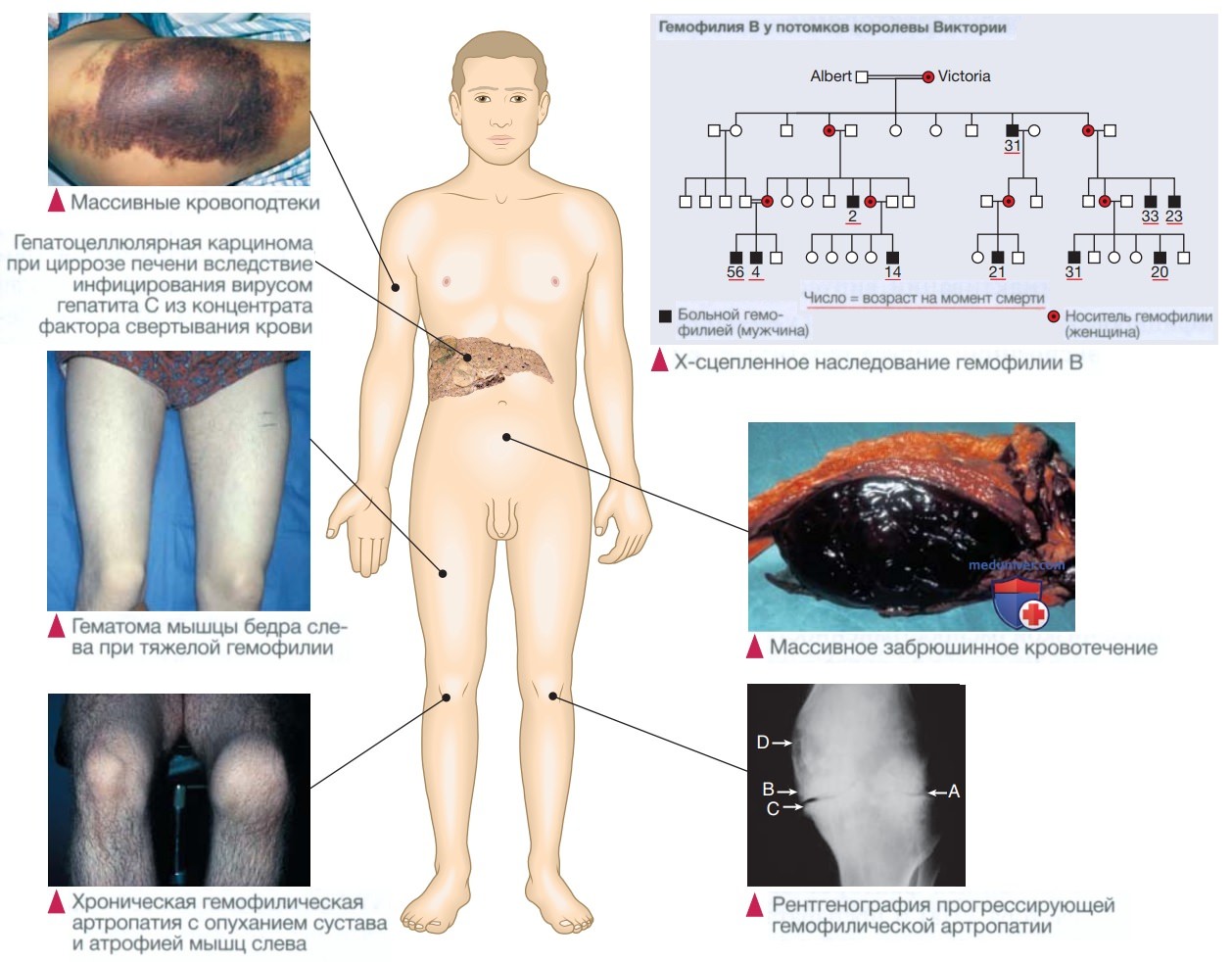
1. Генетика. Ген фактора VIII расположен в одной из Х-хромосом. Гемофилия связана с рядом мутаций в гене фактора VIII; к ним относятся крупные инверсии, большие делеции, миссенс- и нонсенс-мутации, а также мутации сайта сплайсинга. Поскольку ген фактора VIII находится в Х-хромосоме, гемофилия А связана с полом. Таким образом, все дочери пациента с гемофилией являются облигатными носителями гена, и они, в свою очередь, при каждой беременности имеют 1 шанс из 4 родить больного ребенка мужского пола, здорового ребенка мужского пола, девочки-носителя или здоровую девочку. В семьях с выявленной мутацией возможна пренатальная диагностика с помощью биопсии ворсин хориона.
Гемофилия четко наследуется из поколения в поколение; все члены семьи имеют одинаковую мутацию гена фактора VIII и сопоставимый тяжелый или умеренный фенотип. Женщины-носительницы гена гемофилии могут иметь сниженный уровень фактора VIII из-за случайной инактивации их нормальной Х-хромосомы во время внутриутробного развития. Это может привести к развитию легкой коагулопатии; таким образом, у всех известных или предполагаемых носителей гемофилии А следует измерять уровень фактора VIII.
2. Клинические проявления. Степень тяжести и особенности кровотечений тесно связаны с остаточным уровнем фактора VIII (табл. 63). У пациентов с тяжелыми формами гемофилии (концентрация фактора VIII <0,01 Ед/мл) наблюдаются спонтанные кровотечения в кожу, мышцы и суставы. Также характерны забрюшинные и внутричерепные кровоизлияния. Дети с тяжелой гемофилией имеют повышенный риск внутричерепного кровоизлияния, и, несмотря на отсутствие достаточных доказательств, при плановом кесаревом сечении у таких детей необходимо избегать травматизации головы и выполнять нейровизуализацию у новорожденного в течение первых суток после рождения.
Лица с умеренной и легкой гемофилией (концентрация фактора VIII 0,01—0,4 Ед/ мл) имеют аналогичный характер кровотечений, но они обычно возникают после травмы или операции, при этом объем кровотечения существенно превышает тяжесть повреждения.
Вследствие рецидивирующих кровотечений при тяжелой гемофилии чаще всего страдает скелетно-мышечная система. Кровоизлияние обычно происходит в крупные суставы, особенно в коленные, локтевые, голеностопные и тазобедренные. Также характерно развитие мышечных гематом, чаще всего в икроножных и поясничных мышцах. Если сразу же не назначена гемостатическая терапия, то появляется горячая, объемная и очень болезненная гематома в капсуле сустава или в мышечном ложе.
Рецидив кровотечения в сустав приводит к гипертрофии синовиальной оболочки, разрушению хряща и развитию хронической гемофилической артропатии (рис. ниже). Осложнения мышечных гематом зависят от их локализации. Большое кровотечение в поясничную мышцу при распространении может сдавливать бедренный нерв; гематомы мышц голени могут повышать давление в неэластичной фасциальной оболочке, вызывая синдром сдавления с ишемией, некрозом, фиброзом и последующей контрактурой и укорочением ахиллова сухожилия.
Клинические проявления гемофилии. На рентгенограмме коленного сустава повторные кровоизлияния привели к расхождению надмыщелков бедренной кости, а об отсутствии хряща свидетельствует сближение бедренной и большеберцовой кости (А); имеются склероз (В), остеофит (С) и кисты в костной ткани (D). (ВГС — вирус гепатита С) Вклейка (массивные кровоподтеки).
3. Лечение. Основой лечения тяжелой гемофилии А (и В) в развитых странах является профилактическая заместительная терапия фактором свертывания крови. Цель этого лечения — поддержание минимальной концентрации фактора VIII (или IX в случае гемофилии В) выше 0,02 Ед/мл. Это существенно уменьшает количество эпизодов кровотечений у мужчин с тяжелой гемофилией и, следовательно, снижает скорость ухудшения состояния суставов, что является основным отдаленным осложнением.
Профилактику можно назначать различными способами: ежедневно, через день или на основе данных по фармакокинетике, которые позволяют определить лучший способ планирования профилактики.
Клиническая практика лечения гемофилии А и В в настоящее время несколько меняется из-за появления различных концентратов рекомбинантных факторов, обработанных с целью изменения периода полувыведения. В дополнение к рекомбинантному фактору VIII со стандартным периодом полувыведения теперь существуют новые препараты, полученные с помощью Fc-гибридизации и пегилирова-ния/гликопегилирования, увеличивающие период полувыведения фактора VIII до такой степени, что его можно использовать для изменения режимов дозирования с целью профилактики.
Альтернативный подход, который все еще приходится использовать в менее развитых странах, заключается в ситуационном лечении. При тяжелой гемофилии А эпизоды кровотечения следует лечить, повышая концентрацию фактора VIII, как правило, за счет внутривенной инфузии концентрата фактора свертывания крови VIII. Концентраты фактора свертывания крови VIII являются лиофилизированными и стабильными при 4 °C и поэтому могут храниться в бытовых холодильниках, что позволяет пациентам вводить их себе дома самостоятельно при возникновении самых ранних признаков кровотечения. Концентрат фактора свертывания крови VIII, полученный из плазмы крови донора, в настоящее время подвергается скринингу на вирусы гепатитов В и С, а также на ВИЧ и проходит два отдельных процесса инактивации вирусов во время производства; эти препараты имеют хорошие показатели безопасности.
Однако концентраты фактора свертывания крови VIII, полученные с помощью рекомбинантной технологии, в настоящее время широко доступны на рынке и, несмотря на их более высокую стоимость, считаются более безопасными, чем препараты, полученные из плазмы крови человека (в плане риска инфицирования). В дополнение к повышению концентрации фактора свертывания крови VIII иммобилизация зоны кровотечения с помощью постельного режима или наложения шины уменьшает продолжающееся кровотечение. После остановки кровотечения пациент должен быть активизирован, и ему следует назначить физиотерапию для восстановления силы в прилегающих мышцах. Всем неиммунным потенциальным реципиентам объединенных препаратов крови следует предложить иммунизацию против гепатита А и В.
Агонист рецепторов вазопрессина десмопрессин повышает уровни vWF и фактора VIII в 3—4 раза, что помогает при остановке кровотечения у пациентов с легкой или умеренной гемофилией А. Доза, необходимая для этой цели, выше дозы для лечения несахарного диабета (обычно 0,3 мкг/кг), и препарат вводится внутривенно или подкожно. В качестве альтернативы тот же самый эффект можно получить с помощью интраназального введения дозы 300 мкг. После повторного введения десмопрессина у пациентов следует контролировать задержку жидкости, что может привести к существенной гипонатриемии.
Десмопрессин противопоказан пациентам с наличием в анамнезе тяжелого поражения артерий из-за склонности провоцировать тромбозы и маленьким детям, у которых гипонатриемия может привести к судорогам.
4. Осложнения терапии факторами свертывания крови. До 1986 г. концентраты факторов свертывания крови, полученные из плазмы человека, не подвергались процессу инактивации вирусов, и многие пациенты заражались ВИЧ и вирусом гепатита В и С. Пациенты с гемофилией, получавшие объединенные концентраты, которые до 1988 г. не подвергались инактивации вирусов, почти все инфицированы вирусом гепатита С, у 80—90% есть признаки экспозиции вируса гепатита В, а 60% стали ВИЧ-положительными.
Опасение, что возбудитель вариантов болезни Крейтцфельдта—Якоба может передаваться через кровь и препараты крови, было подтверждено у реципиентов эритроцитарной массы и у одного реципиента фактора VIII. Объединенные продукты плазмы, включая концентрат фактора VIII, в настоящее время производятся из плазмы крови, взятой в странах с низкой распространенностью губчатой энцефалопатии крупного рогатого скота.
Еще одним серьезным осложнением при инфузии фактора VIII является выработка против него антител, которые возникают примерно у 20% пациентов с тяжелой гемофилией. Такие антитела быстро нейтрализуют терапевтические инфузии, делая лечение практически неэффективным. Инфузии активированных факторов свертывания (например, фактора VIIa) и препарата, шунтирующего ингибиторы к фактору VIII (Фейба), могут остановить кровотечение.
б) Гемофилия В (болезнь Кристмаса). Аберрации гена фактора IX, который также находится в Х-хромосоме, вызывают снижение концентрации фактора IX в плазме крови, что приводит к развитию гемофилии В. Это заболевание клинически неотличимо от гемофилии А, но встречается реже. Частота эпизодов кровотечения связана с выраженностью дефицита фактора IX.
Для лечения используется концентрат фактора IX, который применяется почти так же, как и фактор VIII при гемофилии А. Новые рекомбинантные препараты фактора IX с увеличенным периодом полувыведения, полученные с помощью Fc-гибридизации, гибридизации с альбумином и пегилирования, дают возможность профилактически вводить их 1 раз в неделю или даже 1 раз в 2 нед.
Несмотря на то что концентраты фактора IX имеют те же проблемы с риском передачи вируса, что и концентраты фактора VIII, они обычно не индуцируют появление ингибиторных антител (<1% пациентов); однако если это происходит, то возникает риск развития тяжелой реакции аллергического типа.
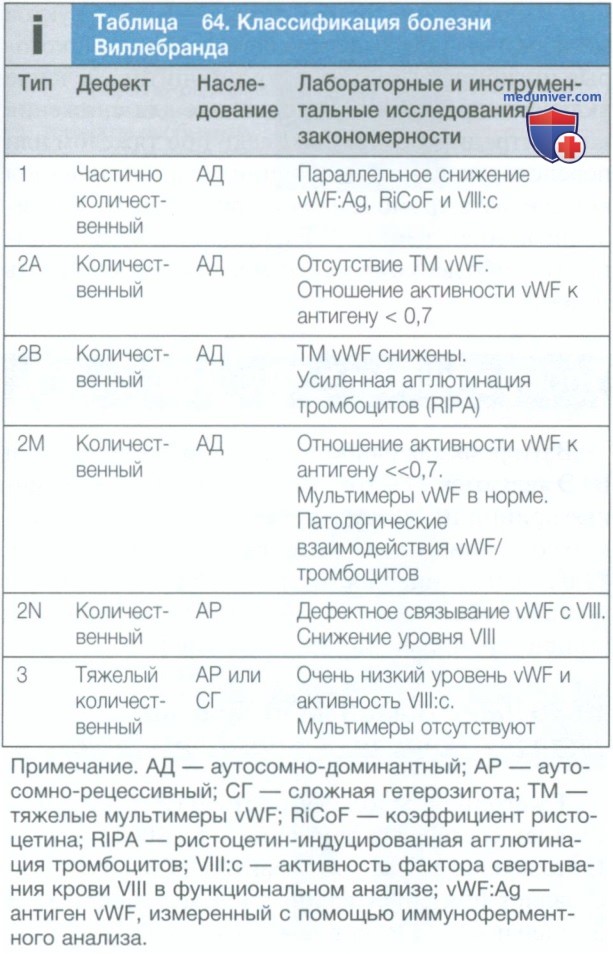
в) Болезнь Виллебранда. Болезнь Виллебранда — распространенное, но обычно легкое нарушение свертывания крови, вызванное количественным (типы 1 и 3) или качественным (тип 2) дефицитом vWF. Этот белок синтезируется эндотелиальными клетками и мегакариоцитами и участвует как в обеспечении функции тромбоцитов, так и в коагуляции.
Обычно он образует полимерную структуру, которая необходима для его взаимодействия с субэндотелиальным коллагеном и тромбоцитами (см. рис. выше). vWF действует как белок-носитель для фактора VIII, с которым он связан нековалентно; дефицит vWF снижает уровень фактора VIII в плазме крови. vWF также образует «мостики» между тромбоцитами и субэндотелиальными компонентами (например, коллагеном; см. рис. 5.6, Б), что позволяет тромбоцитам прилипать к поврежденным стенкам сосудов; следовательно, дефицит vWF приводит к нарушению формирования тромбоцитарного сгустка.
Антигены группы крови (А и В) экспрессируются на vWF, снижая его восприимчивость к протеолизу; в результате у людей с группой крови 0 уровень циркулирующего vWF ниже, чем у людей с другими группами крови. Это необходимо учитывать при постановке диагноза «болезнь Виллебранда».
Большинство пациентов с болезнью Виллебранда имеют вариант 1-го типа, характеризующийся количественным уменьшением нормального функционального белка. Пациенты с нарушением 2-го типа наследуют функционально аномальные молекулы vWF. Тип аномалии зависит от места мутации в гене vWD и от того, как она влияет на связывание с тромбоцитами, коллагеном и фактором VIII. У пациентов с болезнью 2А типа отмечается нарушение vWF-зависимой адгезии тромбоцитов; при мутации в участке связывания гликопротеина IB тромбоцитов, приводящей к увеличению сродства к гликопротеину IB, развивается заболевание 2В типа; при мутации в участке связывания фактора VIII развивается болезнь 2N типа; а при других нарушениях связывания тромбоцитов, но при нормальной полимерной структуре vWF развивается болезнь 2М типа.
Лабораторные изменения, сопровождающие эти типы, описаны в табл. 64. Ген vWF расположен на хромосоме 12, и заболевание обычно наследуется по аутосомно-доминантному типу, за исключением типа 2N и типа 3, где наследование является аутосомно-рецессивным.
1. Клинические проявления. Отмечаются геморрагические проявления, характерные для сниженной функции тромбоцитов. Часто развиваются поверхностные синяки, носовые кровотечения, меноррагии и желудочно-кишечные кровотечения. Эпизоды кровотечения обычно гораздо более редкие, чем при тяжелой гемофилии, а сильные кровотечения могут наблюдаться только после травмы или операции. В пределах одной семьи заболевание имеет различную пенетрантность, поэтому у некоторых членов семьи могут быть довольно тяжелые и частые кровотечения, тогда как у других болезнь протекает относительно бессимптомно.
2. Лабораторные и инструментальные исследования. Заболевание характеризуется сниженной активностью vWF и фактора VIII. Оно может быть диагностировано с помощью набора тестов, которые включают функциональные и антигенные измерения vWF, мультимерный анализ белка и специфические функциональные исследования для определения связывания с гликопротеином Ib тромбоцитов (RIPA) и фактором VIII (табл. 64). Кроме того, в большинстве случаев информативным является анализ мутаций в гене vWF.
3. Лечение. Многие эпизоды небольших кровотечений можно успешно лечить местно или десмопрессином, который повышает уровень vWF, что приводит к вторичному увеличению фактора уровня VIII. Транексамовая кислота может дать эффект при кровотечении из слизистых оболочек. При более серьезных или персистирующих кровотечениях гемостаз может быть достигнут с помощью ряда концентратов фактора VIII, которые содержат значительное количество vWF в дополнение к фактору VIII. Маленькие дети и пациенты с тяжелым поражением артерий не должны получать десмопрессин, а у пациентов с болезнью 2В типа развивается тромбоцитопения, которая может быть нежелательной после введения десмопрессина. При кровотечении у пациентов с болезнью 3-го типа эффективен только концентрат фактора VIII/vWF.
г) Редкие наследственные нарушения свертывания крови. Тяжелые дефициты факторов VII, X и XIII представляют собой аутосомно-рецессивные заболевания. Они встречаются редко, но характеризуются тяжелыми кровотечениями. К типичным проявлениям относят кровотечение из пуповинного остатка и внутричерепное кровоизлияние. Дефицит фактора XIII у женщин обычно связан с рецидивирующей гибелью плода.
Дефицит фактора XI может возникать у гетерозиготных и гомозиготных лиц. Кровотечение может быть различным по интенсивности, и его нельзя точно прогнозировать на основании уровней факторов свертывания. Тяжелые кровотечения в основном возникают у пациентов уровнями факторов свертывания ниже 15% нормы.
д) Приобретенные нарушения свертывания крови. ДВС-синдром является важной причиной кровотечений, которые начинаются с усиленной и неадекватной внутрисосудистой коагуляции. ДВС-синдром обсуждается в разделе «Тромботические заболевания».
1. Заболевания печени. Несмотря на то что традиционно тяжелое паренхиматозное заболевание печени описывается как состояние, связанное с повышенной кровоточивостью, в настоящее время стало очевидным, что у этих пациентов также увеличен риск венозных тромбозов. Несмотря на то что в печени уменьшается синтез прокоагулянтных факторов, это в какой-то степени компенсируется снижением образования естественных антикоагулянтных белков и фибринолитической активности у пациентов с поздними стадиями заболеваний печени. При тяжелых паренхиматозных заболеваниях печени кровотечение может возникать по разным причинам.
Часто присутствуют патологические источники потенциального массивного кровотечения, такие как варикозное расширение вен пищевода или пептическая язва. Снижается синтез белков в печени, таких как факторы V, VII, VIII, IX, X, XI, протромбин и фибриноген. Клиренс активатора плазминогена уменьшается. Может возникать тромбоцитопения вследствие гиперспленизма при портальной гипертензии. При холестатической желтухе ухудшается всасывание витамина К, что приводит к дефициту факторов II, VII, IX и X, а также протеинов С и S. Лечение препаратами плазмы или переливание тромбоцитарной массы следует проводить при острых кровотечениях и при выполнении интервенционных процедур, таких как биопсия печени.
Дефицит витамина К легко корректируется парентеральным введением витамина К (в РФ не зарегистрирован).
2. Почечная недостаточность. Тяжесть кровотечения при почечной недостаточности пропорциональна концентрации мочевины в плазме крови. Кровотечение обусловлено дисфункцией тромбоцитов, при этом желудочно-кишечные кровотечения развиваются особенно часто. Причины обычно многофакторные и включают анемию, небольшую тромбоцитопению и накопление низкомолекулярных продуктов метаболизма, обычно выделяемых почками, которые ингибируют функцию тромбоцитов. Лечение включает проведение гемодиализа для снижения концентрации мочевины. Редко при тяжелом или персистирующем кровотечении показаны инфузии концентрата тромбоцитов и эритроцитов. Увеличение концентрации vWF с помощью криопреципитата либо десмопрессина может способствовать гемостазу.
Видео схема гемостаза - механизмы внешнего и внутреннего пути