Синдром Клайнфельтера - кратко с точки зрения внутренних болезней
Синдром Клайнфельтера встречается примерно у одного из 1000 мужчин и обычно связан с кариотипом 47XXY. Однако возможны и другие цитогенетические варианты, особенно мозаичная форма 46XY/47XXY. Основным патологическим нарушением является дисгенезия семенных канальцев.
Ее можно обнаружить с младенчества (и, возможно, даже внутриутробно), и с возрастом дисгенезия канальцев прогрессирует. К подростковому возрасту в семенных канальцах развиваются гиалинизация и фиброз, нарушается функция клеток Лейдига, что приводит к гипогонадизму.
а) Клиническая картина. Диагноз обычно ставится у подростков с гинекомастией и задержкой полового созревания. У этих пациентов обычно маленькие, твердые яички. Высокий рост заметен с раннего детства, характерны длинные ноги, что связано с кариотипом 47XXY, и может усугубляться дефицитом андрогенов с отсутствием закрытия эпифизарных зон роста в период полового созревания.
Другие клинические проявления могут включать сложности в обучении и наличие поведенческих расстройств, а также повышенный риск развития рака молочной железы и сахарного диабета 2-го типа в более старшем возрасте. Спектр клинических характеристик широк, и у некоторых людей, особенно с мозаичной формой 46XY/47XXY, половое созревание может проходить нормально, а диагноз ставится только во время обследования по поводу бесплодия.
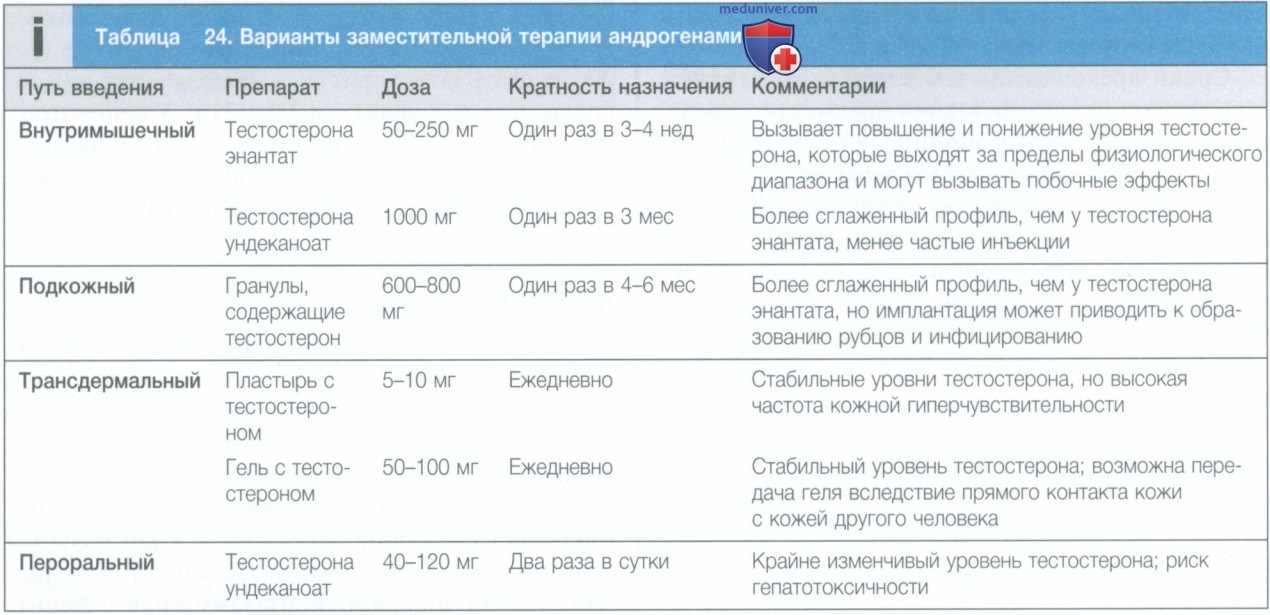
б) Диагностика и лечение. Синдром Клайнфельтера можно заподозрить по типичному фенотипу у пациента с гипергонадотропным гипогонадизмом и подтвердить с помощью кариотипирования. Лицам с клиническими и биохимическими признаками дефицита андрогенов требуется заместительная терапия андрогенами (см. табл. 24).
Описаны случаи успешного наступления беременности после терапии интрацитоплазматической инъекцией сперматозоидов, когда сперматоциты были извлечены из половых желез мужчин с синдромом Клайнфельтера.