Иммунодефицит - кратко с точки зрения внутренних болезней
Последствия иммунодефицита включают рецидивирующие инфекции, аутоиммунные реакции (как результат нарушения регуляции иммунной системы) и повышенную предрасположенность к злокачественным новообразованиям, особенно к ассоциированным с вирусными инфекциями, такими как ВЭБ.
Иммунодефицит может возникать из-за внутренних дефектов иммунной системы, но гораздо чаще обусловлен вторичными причинами, включая инфекции, последствия фармакотерапии, злокачественные новообразования и пожилой возраст. В этом разделе приведен обзор первичных иммунодефицитных состояний.
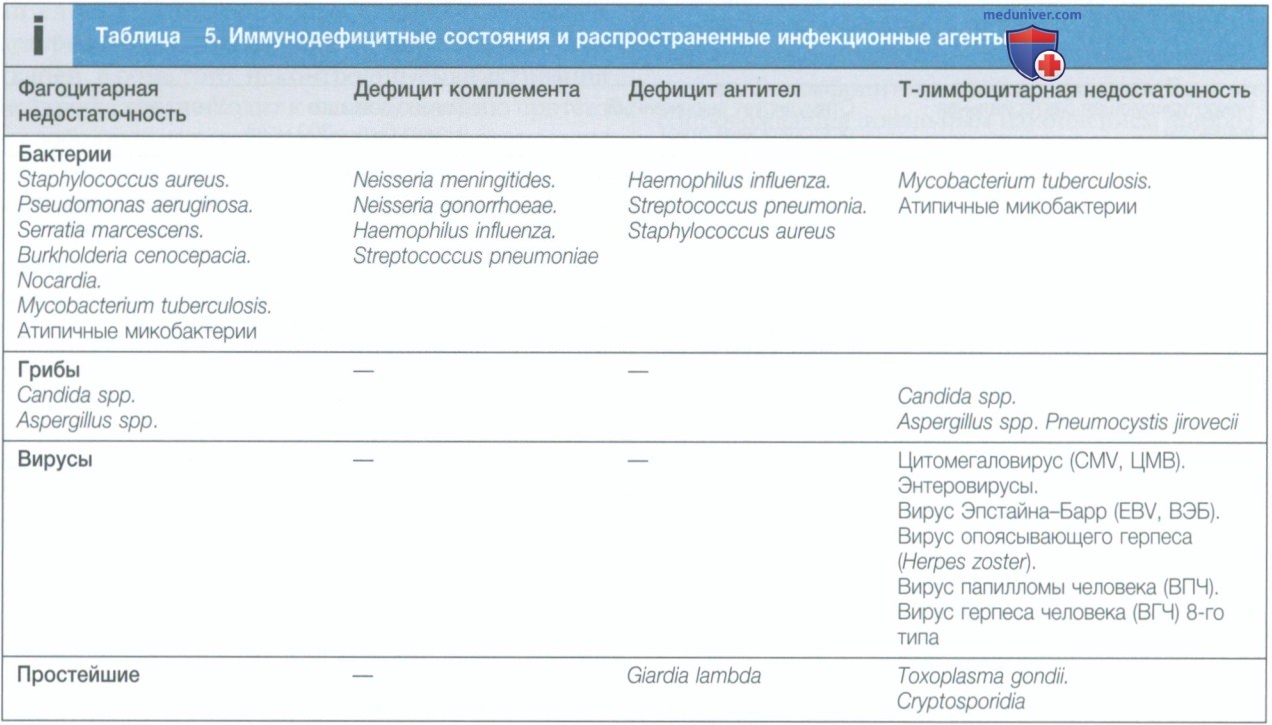
На сегодняшний день описано более 100 таких иммунодефицитов, большинство из которых генетически обусловлены и манифестируют в детстве или подростковом возрасте. Клиническая картина зависит от того, какой компонент иммунной системы поврежден (см. табл. 5). Однако иммунные механизмы в значительной степени перекрывают и дублируют друг друга, поэтому некоторые заболевания не всегда в полной мере соответствуют данной классификации.
а) Первичная недостаточность фагоцитов. Первичная недостаточность фагоцитов обычно проявляется рецидивирующими бактериальными и грибковыми инфекциями, нередко необычной локализации. Таким пациентам требуется активное лечение инфекции, включая в/в введение антибиотиков и хирургическое дренирование абсцессов, а также долгосрочная профилактика с помощью антибактериальных и противогрибковых препаратов.
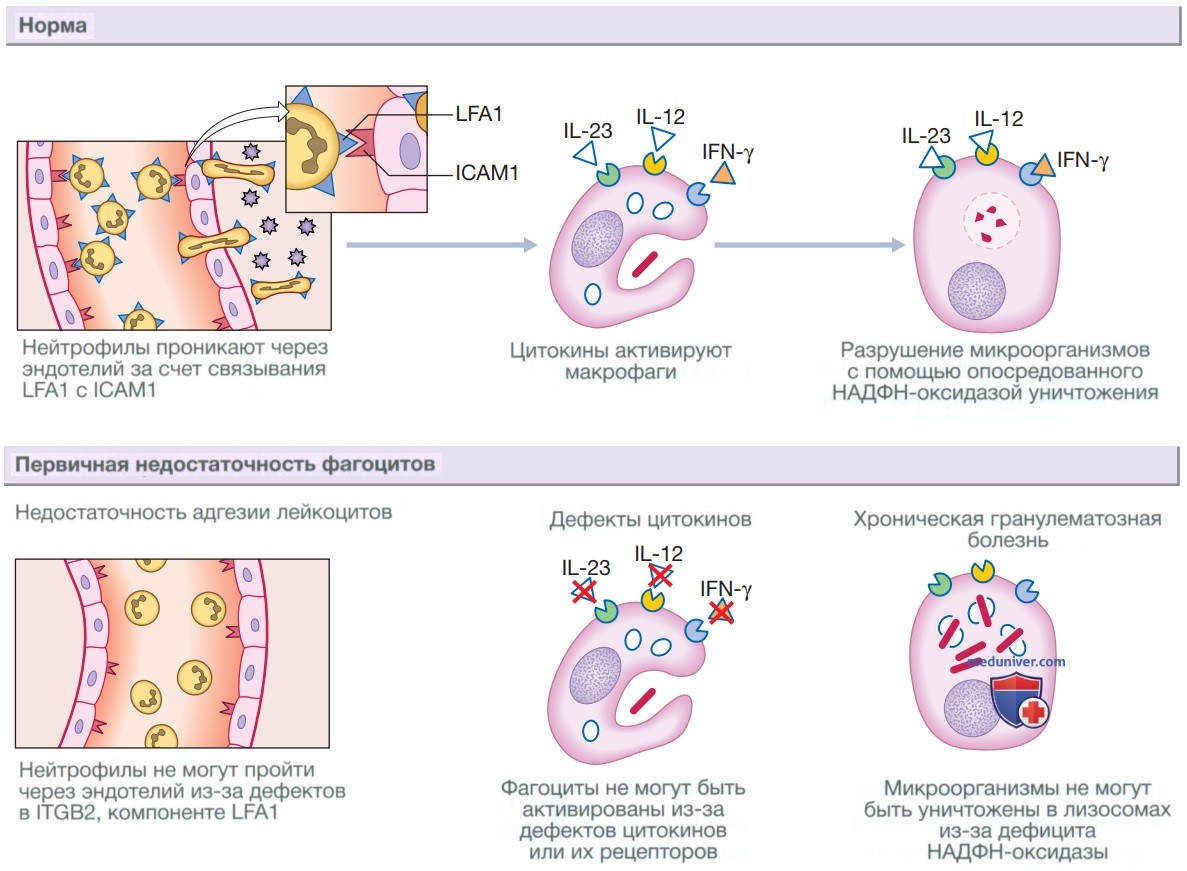
Основные варианты первичной недостаточности фагоцитов представлены на рис. 1 и обсуждаются ниже.
Рисунок 1. Функция фагоцитов в норме и механизмы первичной недостаточности фагоцитов. В норме нейтрофилы проходят через эндотелий и проникают в ткани с помощью молекулы, локализованной на их мембране, — функционального антигена 1 лимфоцитов (Lymphocyte Function-Associated antigen 1 — LFA1), который связывается с молекулой межклеточной адгезии 1-го типа (Intercellular Adhesion Molecule 1 — ICAM1), расположенной на эндотелии. Для того чтобы макрофаги могли поглотить и уничтожить микроорганизмы, они должны быть активированы цитокинами, а также для образования свободных радикалов им требуется наличие никотинамидадениндинуклеотидфосфатоксидазы. Первичная недостаточность фагоцитов может возникать в результате неспособности лейкоцитов проникать через эндотелий из-за дефектов LFA1 вследствие мутаций в генах, ответственных за синтез цитокинов или их рецепторов, а также дефектов НАДФН-оксидазы. γ-ИФН — γ-интерферон; ИЛ — интерлейкин
1. Хроническая гранулематозная болезнь. Хроническая гранулематозная болезнь вызвана мутациями в генах, кодирующих ферменты НАДФН-оксидазы, что приводит к нарушению механизма кислородзависимого уничтожения клеток (киллинга). Этот дефект повышает восприимчивость к инфекциям, вызванным каталазоположительными микроорганизмами, такими как Staphylococcus aureus, Burkholderia cenocepacia и Aspergillus.
Кроме того, нарушается процесс внутриклеточного киллинга в фагоцитах. Инфекционный процесс чаще всего затрагивает легкие, лимфатические узлы, мягкие ткани, кости, кожу и мочевыводящие пути, а гистологически данный процесс характеризуется формированием гранулем. Большинство случаев заболевания сцеплены с Х-хромосомой.
2. Недостаточность адгезии лейкоцитов. К недостаточности (дефициту) адгезии лейкоцитов относят очень редкие виды нарушений миграции фагоцитов, возникающие вследствие недостаточной экспрессии молекул адгезии на поверхности лейкоцитов, что приводит к их неспособности покидать кровоток. Наиболее частой причиной являются мутации, приводящие к утрате функции гена ITGB2, который кодирует синтез β2-цепи интегрина, компонента молекулы адгезии LFA1.
Они характеризуются рецидивирующими бактериальными инфекциями, но в очагах ее локализации отсутствуют признаки нейтрофильной инфильтрации, например образование гноя. Количество нейтрофилов в периферической крови во время острой фазы инфекционного процесса может быть очень высоким из-за неспособности мобилизованных нейтрофилов выйти за пределы кровеносных сосудов. Специальные тесты выявляют снижение или отсутствие экспрессии молекул адгезии на нейтрофилах.
3. Дефекты цитокинов и рецепторов цитокинов. Мутации генов, кодирующих такие цитокины, как γ-ИФН, ИЛ-12, ИЛ-23, или их рецепторы, приводят к нарушению внутриклеточного кил-линга в макрофагах, и такие пациенты становятся особенно чувствительными к микобактериальным инфекциям.
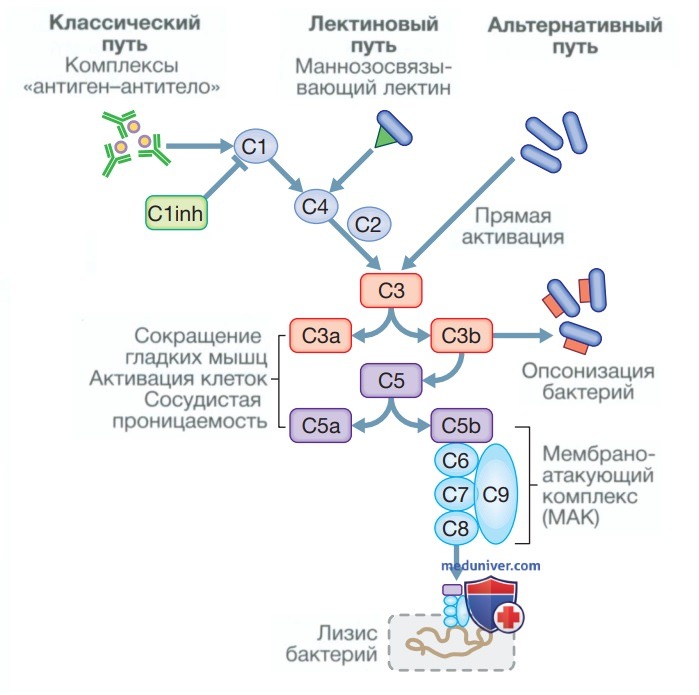
б) Первичные иммунодефициты с дефектом в системе комплемента. Мутации с потерей функции были описаны почти для всех белков системы комплемента (см. рис. ниже). В то время как большинство из них встречаются редко, дефицит маннозосвязывающего лектина широко распространен и имеется примерно у 5% населения Северной Европы, в большинстве случаев протекая бессимптомно.
Путь комплемента. Классический путь активируется связыванием комплексов «антиген-антитело» с С1, а блокируется ингибитором С1 (С1 inhibitor — С1 inh), тогда как маннозосвязывающие лектины, представляющие собой макромолекулы, связывающиеся с различными микроорганизмами, активируют этот путь при связывании С4. Бактерии могут напрямую активировать путь через С3, который играет ключевую роль в активации комплемента всеми тремя путями
1. Клиническая картина. Недостаточность белков системы комплемента может манифестировать по-разному. В некоторых случаях она проявляется рецидивирующими инфекциями, вызванными инкапсулированными бактериями (например, Neisseria spp.), что отражает важность мембраноатакующего комплекса в защите от микроорганизмов данного вида. Кроме того, генетические дефекты компонентов классического пути активации комплемента (C1, С2 и С4) повышают риск развития аутоиммунных заболеваний, в частности СКВ.
У лиц с дефицитом маннозосвязывающего лектина высока заболеваемость бактериальными инфекциями при наличии других иммунокомпрометирующих состояний, таких как недоношенность или проводимая химиотерапия. Тем не менее значимость данной патологии остается дискутабельной, поскольку популяционные исследования не выявили общего увеличения частоты инфекционных заболеваний и общей смертности у таких пациентов.
Дефицит регуляторного белка ингибитора С1 не сопровождается рецидивирующими инфекциями, но провоцирует повторные ангионевротические отеки.
2. Лабораторные и инструментальные исследования. Скрининг на выявление недостаточности белков системы комплемента обычно включает специальные функциональные тесты, основанные на комплемент-опосредованном гемолизе. К ним относят тесты СН50 (классический гемолитический путь активации комплемента 50) и АР50 (альтернативный путь активации комплемента 50). В случае выявления отклонений от нормы результатов скрининговых гемолитических тестов проводится оценка активности компонентов комплемента в отдельности.
3. Лечение. Пациентов с дефицитом белков системы комплемента необходимо вакцинировать от менингококка, пневмококка и Н. influenzae типа В для активации приобретенного иммунитета. Кроме того, им рекомендуется пожизненное профилактическое назначение пенициллина для предотвращения развития менингококковой инфекции, а в случае любого инфекционного заболевания — незамедлительное обращение за медицинской помощью.
Пациентам также следует носить MedicAlert или аналогичный браслет. У членов семей пациентов, входящих в группу риска, необходимо проводить скрининговое обследование на предмет выявления дефицита системы комплемента с помощью функциональных скрининговых тестов. Лечение дефицита С1-эстеразы обсуждается в другом разделе.
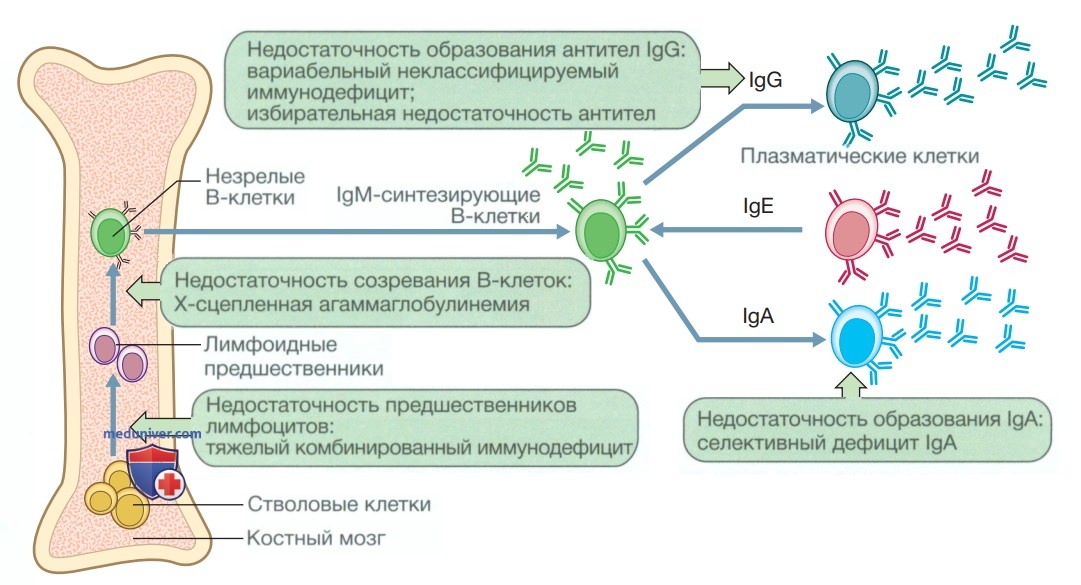
в) Первичные иммунодефициты с преимущественной недостаточностью антител. Первичные иммунодефициты с преимущественной недостаточностью антител формируются в результате аномального функционирования В-клеток, как показано на рис. 2. Они характеризуются рецидивирующими бактериальными инфекциями, особенно дыхательных путей и желудочно-кишечного тракта. Наиболее частыми инфекционными агентами являются инкапсулированные бактерии, такие как Streptococcus pneumoniae и Н. influenzae.
Рисунок 2. В-клетки и виды первичных иммунодефицитов с преимущественной недостаточностью антител (зеленые блоки). Ig — иммуноглобулин
Эти нарушения обычно манифестируют в младенческом возрасте, когда ослабевает защитное действие полученных трансплацентарно материнских Ig. Ниже обсуждаются наиболее важные причины возникновения первичных иммунодефицитов с преимущественной недостаточностью антител.
1. Сцепленная с Х-хромосомой агаммаглобулинемия (Брутона). Это редкое заболевание с Х-сцепленным типом наследования, возникающее вследствие мутаций в гене ВТК, который отвечает за синтез тирозин-киназы Брутона (Bruton) — сигнального белка, необходимого для созревания В-клеток. Страдают лица мужского пола, которые с младенчества подвержены тяжелым бактериальным инфекциям. При данном заболевании наблюдается значительное уменьшение числа В-клеток, а уровни Ig снижены либо не определяются.
Лечение сцепленной с Х-хромосомой агаммаглобулинемии состоит в заместительной терапии Ig и назначении антибиотиков для борьбы с инфекционными осложнениями.
2. Избирательный дефицит иммуноглобулина А. Это самый часто встречающийся из первичных иммунодефицитов с преимущественной недостаточностью антител, заболеваемость которым среди жителей Северной Европы составляет 1 на 600 населения. Несмотря на то что дефицит IgA обычно протекает бессимптомно и не имеет клинических осложнений, примерно 30% пациентов с данной патологией подвержены рецидивирующим респираторным и желудочно-кишечным заболеваниям, протекающим в легкой форме.
Диагноз подтверждается при определении низких уровней IgA или при невозможности их выявления (менее 0,05 г/л). У некоторых пациентов имеется компенсаторное повышение в сыворотке уровня IgG. Как правило, специфического лечения не требуется.
3. Общий вариабельный иммунодефицит. Общий вариабельный иммунодефицит / общая вариабельная иммунная недостаточность характеризуется снижением сывороточного IgG и недостаточной продукцией антител в ответ на воздействие экзогенных патогенов. Это гетерогенный первичный иммунодефицит, манифестирующий во взрослом возрасте, причина которого неизвестна. Клинически заболевание проявляется рецидивирующими инфекциями с развитием осложнений в виде формирования бронхоэктазов.
Как ни парадоксально, но для общей вариабельной иммунной недостаточности типично возникновение аутоиммунных заболеваний с антителоопосредованным механизмом развития, таких как идиопатическая тромбоцитопеническая пурпура и аутоиммунная гемолитическая анемия. Кроме этого, общая вариабельная иммунная недостаточность также ассоциирована с повышенным риском развития злокачественных новообразований, в особенности лимфопролиферативных заболеваний.
4. Функциональный дефицит антител иммуноглобулина G*. Это недостаточно изученное состояние, которое характеризуется нарушением специфического гуморального ответа на контакт с полисахаридными антигенами. Кроме того, у ряда пациентов также выявляется селективный дефицит IgG2 и IgG4, в связи с чем ранее данную патологию относили к избирательному дефициту подклассов IgG. Специфический дефицит антител, селективный дефицит IgA и общая вариабельная иммунная недостаточность имеют некоторые общие черты, и с течением времени у отдельных пациентов эти состояния могут привести к тотальной недостаточности антителообразования.
P.S. *Синоним, используемый далее в тексте, — специфический дефицит антител.
5. Лабораторные и инструментальные исследования. Для исключения вторичного характера гипогаммаглобулинемии следует определять содержание сывороточных Ig (табл. 15) в сочетании с электрофоретическим исследованием белков сыворотки крови и мочи. Кроме этого, показано типирование субпопуляций Т- и В-лимфоцитов. Оценку специфического гуморального ответа проводят, определяя уровень IgG к известным патогенам — возбудителю столбняка, Н. influenzae и S. pneumoniae (у большинства пациентов в анамнезе можно выявить контакт с данными патогенами вследствие перенесенной инфекции либо после специфической иммунизации). При низком уровне специфических антител необходимо вакцинировать пациента соответствующей убитой вакциной с последующим повторным определением уровня антител через 6—8 нед; отсутствие ответа свидетельствует о значимом дефекте антителообразования.
Данные функциональные тесты в целом заменили количественную оценку подклассов IgG.
6. Лечение. Пациенты с иммунодефицитами с преимущественной недостаточностью антителообразования, как правило, нуждаются в активном лечении инфекций и профилактическом назначении антибиотиков. Исключением является избирательный дефицит IgA, который не нуждается в терапии. Основой лечения для подавляющего большинства таких пациентов является заместительная терапия Ig. Препарат Ig получают из плазмы крови сотен доноров, поэтому он содержит антитела класса G к широкому диапазону распространенных микроорганизмов. При заместительной терапии Ig вводится в/в или подкожно, а целью лечения является поддержание его минимальной остаточной концентрации (концентрация IgG непосредственно перед очередным его введением) в пределах нормальных значений.
Такая тактика ведения пациентов минимизирует поражение органов-мишеней и улучшает клинические исходы. Пациенты могут самостоятельно вводить себе Ig, но лечение должно проводиться пожизненно. Вследствие нарушения синтеза IgG эффективность иммунизации у данных пациентов невысока, и, как и у всех пациентов с первичными иммунодефицитами с преимущественным дефектом антителообразования, следует избегать применения живых вакцин.
г) Первичные иммунодефициты с преимущественной недостаточностью Т-лимфоцитов. Данная группа заболеваний характеризуется рецидивирующими инфекциями вирусной, протозойной и грибковой этиологии (см. табл. 5). Кроме того, многие иммунодефициты с преимущественной недостаточностью Т-клеток сопровождаются нарушением антителообразования, поскольку этим клеткам принадлежит важная роль в регулировании функционирования В-клеток. Эта патология обычно проявляется в детстве. Описано несколько причин развития дефицита Т-клеток, которые проиллюстрированы на рис. 3 и ниже обсуждаются детально.
Рисунок 3. Функционирование Т-лимфоцитов в норме и при патологии (зеленые блоки). HLA — главный комплекс гистосовместимости
1. Синдром ДиДжорджи. Этот синдром обусловлен нарушением развития третьей и четвертой жаберных дуг, причиной которого обычно является делеция в хромосоме 22q1. Считается, что иммунодефицит развивается как следствие недоразвития тимуса, однако характер иммунных нарушений может быть очень разнообразным. Для пациентов, страдающих этим заболеванием, характерно очень низкое число циркулирующих в периферической крови Т-клеток, несмотря на их нормальное развитие в костном мозге. Обычно при синдроме ДиДжорджи имеются множественные аномалии развития, такие как врожденные пороки сердца, гипопаратиреоз, трахеопищеводные свищи, заячья губа и волчья пасть.
2. Синдромы голых лимфоцитов (синдром Луи-Бар). Эти редкие состояния возникают вследствие мутаций различных генов, регулирующих экспрессию или транспортировку молекул HLA на поверхность Т-лимфоцитов. Так, например, при патологии молекул HLA I класса нарушается нормальное развитие Т-супрессоров (CD8+), в то время как отсутствие экспрессии молекул HLA II класса приводит к нарушению созревания Т-хелперов (CD4+). Помимо рецидивирующих инфекций, нарушение экспрессии молекул HLA I класса может проявляться системными васкулитами вследствие неконтролируемой активации NK-клеток.
3. Тяжелая комбинированная иммунная недостаточность. Тяжелая комбинированная иммунная недостаточность возникает в результате мутаций в генах, осуществляющих регуляцию лимфогенеза, вследствие чего угнетается созревание Т-клеток. При этом сопутствующее подавление роста и развития В- и NK-клеток также может иметь место, а может отсутствовать. Самой частой причиной развития тяжелой комбинированной иммунной недостаточности является наследуемая сцепленно с Х-хромосомой инактивирующая мутация в гене, кодирующем γ-цепь рецептора к ИЛ-2 (IL2RG), которая является структурным компонентом нескольких интерлейкиновых рецепторов, включая ИЛ-2, ИЛ-7 и ИЛ-15, необходимых для созревания Т- и NK-клеток.
Таким образом, в результате данной мутации формируется тяжелая комбинированная иммунная недостаточность с подавлением этих субпопуляций лимфоцитов: отсутствуют Т- и NK-клетки, присутствуют В-клетки. Еще одной причиной этого вида иммунодефицита является дефицит фермента аденозиндезаминазы (ADA), который приводит к гибели лимфоцитов вследствие внутриклеточного накопления в них токсичных пуриновых метаболитов с последующим развитием тяжелой комбинированной иммунной недостаточности и тотальным угнетением всех субпопуляций лимфоцитов: Т-, NK- и В-клеток.
Отсутствие эффективного адаптивного иммунного ответа способствует развитию вскоре после рождения рецидивирующих бактериальных, грибковых и вирусных инфекций. Трансплантация костного мозга является оптимальным вариантом лечения таких пациентов. Для лечения дефицита ADA при отсутствии подходящего донора для трансплантации костного мозга была одобрена генная терапия, возможности которой в настоящее время являются объектом исследований в отношении тяжелой комбинированной иммунной недостаточности, развившейся вследствие других причин.
4. Лабораторные и инструментальные исследования. Основным лабораторным тестом для выявления нарушений в Т-лимфоцитарном звене является подсчет общего числа лимфоцитов и отдельных субпопуляций лимфоцитов. Кроме этого, показано определение уровня Ig в сыворотке крови. Следующий этап обследования таких пациентов включает проведение функциональных тестов для оценки способности Т-клеток к активации и пролиферации. Пациенты, у которых подозревается недостаточность Т-лимфоцитарного звена иммунитета, должны быть обследованы на наличие ВИЧ-инфекции.
5. Лечение. У пациентов с иммунодефицитами с преимущественной недостаточностью Т-клеточного звена следует проводить профилактику пневмоцистной и грибковой инфекций, а при их возникновении необходима активная лечебная тактика. При сопутствующем дефиците антителообразования показано назначение заместительной терапии Ig. При ряде патологий могут потребоваться трансплантация стволовых клеток или генная терапия. При отягощенном семейном анамнезе и подтвержденном результатами антенатального обследования дефекте Т-клеточного звена иммунитета начатая до развития рецидивирующей инфекции терапия стволовыми клетками может улучшить прогноз.
д) Аутоиммунный лимфопролиферативный синдром. Это редкое заболевание развивается вследствие нарушения естественного апоптоза лимфоцитов, самой частой причиной которого является наличие мутаций в гене FAS, кодирующем синтез одноименного белка. Fas-сигнальный белок принимает участие в регуляции запрограммированной гибели клеток в лимфоцитах. Нарушение Fas-опосредованного апоптоза приводит к массивному накоплению аутореактивных Т-клеток, что в дальнейшем становится причиной развития аутоиммуноопосредованной анемии, тромбоцитопении и нейтропении. Другими клиническими проявлениями этого заболевания являются лимфаденопатия, спленомегалия и целый ряд других аутоиммунных нарушений. Кроме того, на фоне нейтропении повышается восприимчивость к инфекционным заболеваниям.
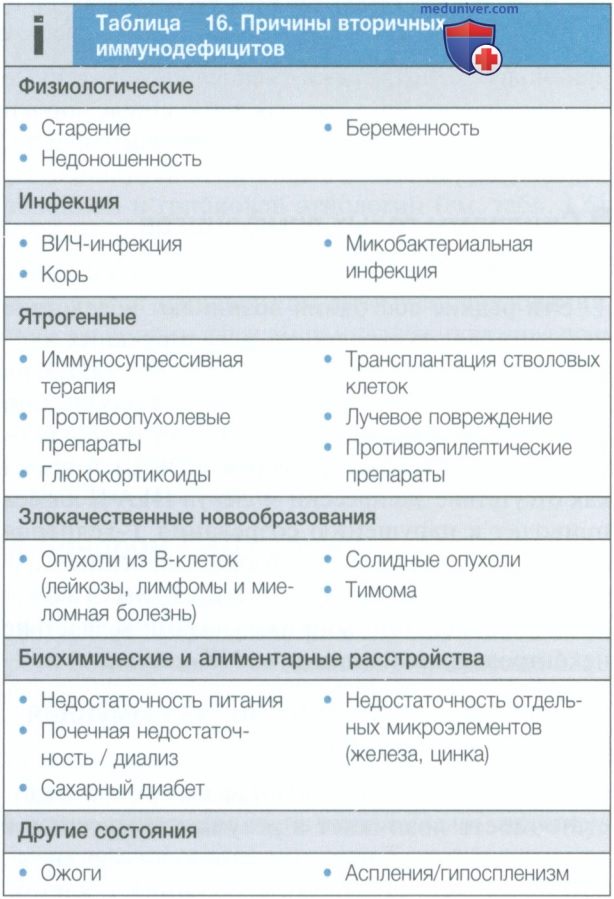
е) Вторичные иммунодефициты. Вторичные иммунодефициты встречаются гораздо чаще, чем первичные, и возникают вследствие воздействия на иммунную систему внешних факторов (табл. 16). Наиболее частыми причинами являются инфекции, такие как ВИЧ и корь, цитотоксические и иммуносупрессивные препараты (особенно это относится к препаратам, которые используются в трансплантологии, а также при лечении онкологических и аутоиммунных заболеваний). Физиологический иммунодефицит встречается в начале и в конце жизни.
Снижение иммунного ответа в пожилом возрасте известно как иммунное старение (табл. 17). Лечение вторичных иммунодефицитов описано в соответствующих главах, посвященных инфекционным болезням, ВИЧ, болезням органов кроветворения и онкологическим заболеваниям.