Болезнь Коновалова—Вильсона (гепатолентикулярная дегенерация) - кратко с точки зрения внутренних болезней
Болезнь Коновалова—Вильсона (гепатолентикулярная дегенерация) — редкое, но значимое нарушение обмена меди, вызванное различными мутациями гена АТР7В, локализованного в хромосоме 13 и наследуемое по аутосомно-рецессивному типу. При этой патологии в организме увеличивается общее количество меди, и ее избыток откладывается в некоторых органах, вызывая их поражение.
а) Патофизиология. Обычно потребляемая с пищей медь всасывается из желудка и проксимального отдела тонкой кишки и быстро попадает в печень, где запасается и включается в церулоплазмин, выделяемый в кровь.
При болезни Коновалова—Вильсона почти всегда происходит нарушение синтеза церулоплазмина, но этот дефект не является первичным, так как около 5% пациентов имеют нормальный уровень церулоплазмина. При рождении содержание меди в организме нормальное, но в дальнейшем оно неуклонно растет; в наибольшей степени от этого страдают печень, базальные ганглии головного мозга, глаза, почки и кости.
Накопление избыточного количества меди в организме в норме предотвращается ее выделением преимущественно с желчью.
Ген АТР7В кодирует белок из семейства аденозинтрифосфатаз P-типа, который выводит медь из клеток различных типов. Было описано не менее 200 различных мутаций. В большинстве случаев это сложные гетерозиготы с двумя разными мутациями гена ATP7B. Попытки соотнести генотип с клинической картиной и течением не выявили каких-либо воспроизводимых закономерностей.
Вследствие большого числа причинных мутаций генетическая диагностика болезни Коновалова—Вильсона, в отличие от гемохроматоза, не является рутинной, хотя она может играть определенную роль в скрининге родственников после идентификации генотипа пробанда.
б) Клиническая картина. Симптомы обычно возникают в возрасте от 5 до 45 лет. Поражение печени встречается преимущественно в детском и раннем подростковом возрасте, хотя оно может проявляться и после 50 лет. Поражение нервной системы проявляется симптомами вовлеченности базальных ганглиев и деменцией, что обычно проявляется в более позднем подростковом возрасте. Проявления могут возникать по отдельности или одновременно. Реже встречаются поражение почечных канальцев и остеопороз.
1. Поражение печени. Возможны эпизоды острого гепатита, иногда рецидивирующие, особенно у детей, также возможно развитие фульминантной печеночной недостаточности, при которой высвобождение свободной меди в кровоток приводит к массивному гемолизу и тубулопатии. Возможен хронический гепатит, который незаметно приводит к циррозу печени, печеночной недостаточности и портальной гипертензии.
Вероятность болезни Коновалова—Вильсона должна быть рассмотрена у любого пациента моложе 40 лет с рецидивирующим острым или хроническим гепатитом неизвестной этиологии, особенно если они сопровождаются гемолизом.
2. Неврологические нарушения. Клиническая картина включает множество экстрапирамидных симптомов (в частности, тремор, хореоатетоз, дистонию, паркинсонизм) и деменцию. Ранним симптомом может быть необычная для возраста неуклюжесть. Неврологическая симптоматика обычно развивается после поражения печени и может быть предотвращена путем эффективного лечения после распознавания болезни на ранних стадиях. Это повышает важность диагностики на этапе поражения печени.
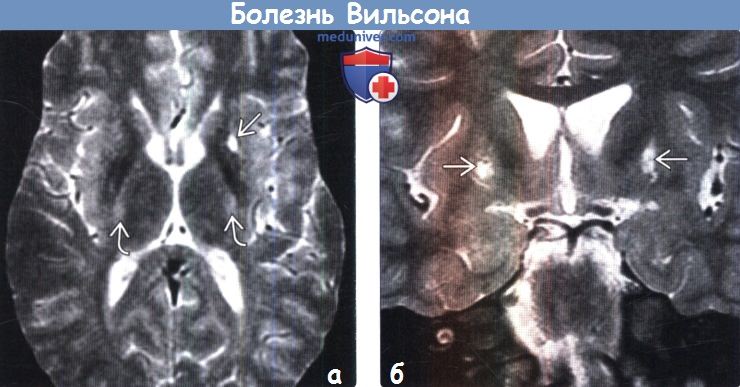
(а) МРТ, Т2-ВИ, аксиальный срез: у мужчины 19 лет в передних отделах скорлупы, а также по ходу кортикоспинальных трактов в задних бедрах внутренних капсул определяются ламинарные гиперинтенсивные зоны.
(б) МРТ, Т2-ВИ, корональный срез: у этого же пациента в скорлупе билатерально визуализируются гиперинтенсивные очаги поражения.
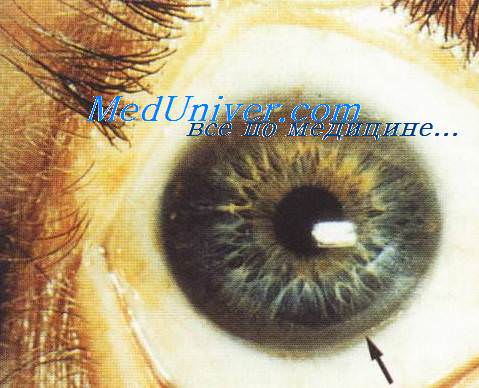
3. Кольца Кайзера-Флейшера. Кольца Кайзера—Флейшера представляют собой зеленовато-коричневые прокрашивания периферии роговицы, изначально заметные в верхней ее части. Это наиболее важный симптом, наличие которого позволяет поставить диагноз. Они обнаруживаются у 60% взрослых с болезнью Коновалова—Вильсона, реже у детей, и почти всегда при неврологических проявлениях заболевания. Иногда их можно выявить только при осмотре с помощью щелевой лампы. На фоне лечения кольца Кайзера—Флейшера исчезают.
Кольцо Кайзера-Флейшера при болезни Вильсона-Коновалова
в) Диагностика. Низкий уровень церулоплазмина в сыворотке — лучший лабораторный показатель, позволяющий поставить диагноз. Однако уровень церулоплазмина снижается при прогрессировании печеночной недостаточности независимо от ее причины, а при болезни Коновалова—Вильсона он иногда не снижается. Именно поэтому необходимо исследовать другие показатели нарушения обмена меди; к ним относят высокую концентрацию свободной меди в сыворотке, повышение экскреции меди с мочой более 0,6 мкмоль (38 мкг) в сутки и очень высокое содержание меди в печени.
Экскреция меди более 25 мкмоль/сут на фоне терапии D-пеницилламином подтверждает диагноз.
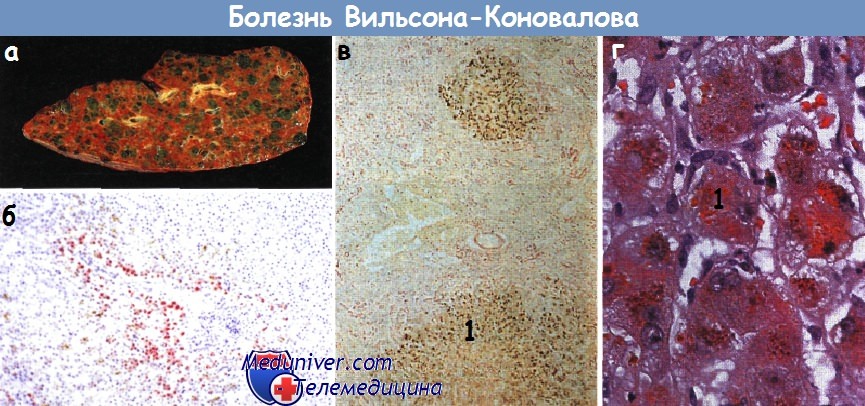
а - Болезнь Вильсона-Коновалова. В препарате после гепатэктомии виден крупноузловой цирроз.
б - Микроскопическая картина при болезни Вильсона-Коновалова, окраска на выявление меди.
Красно-коричневые отложения меди в перипортальных гепатоцитах.
в,г - Гистологическая картина печени при болезни Вильсона-Коновалова. Отложения меди в соединении с белком.
Окраска рубеановой кислотой (а) (х 20). Окраска гематоксилин-эозином (б) (х 320). 1 — белково-медный комплекс
г) Лечение. Препаратом выбора является D-пеницилламин, который связывает медь. Назначенная доза должна быть достаточной для усиления выведения меди с мочой, большинству пациентов требуется доза 1,5 мкг/сут (1—4 мкг/сут). Доза может быть снижена при достижении ремиссии, но лечение должно продолжаться всю жизнь, даже во время беременности, чтобы избежать повторного накопления меди. Резкое прекращение лечения может вызвать острую печеночную недостаточность.
У 1/3 пациентов возникают токсические эффекты: сыпь, нефропатия с протеинурией, волчаночноподобный синдром и миелосупрессия. В случае их развития альтернативой являются триентина дигидрохлорид (1,2—2,4 мкг/сут) и цинк (50 мг 3 раза в сутки).
Трансплантация печени показана при печеночной недостаточности (фульминантной или на фоне цирроза). Значение трансплантации печени при тяжелой неврологической симптоматике не ясно. Прогноз очень хороший при условии, что лечение начато до появления необратимых изменений. Братья, сестры и дети пациентов с болезнью Коновалова-Вильсона должны быть обследованы, и в случае подтверждения диагноза лечение должно проводиться даже при отсутствии симптомов.