Врожденные дизэритропоэтические анемии (ДЭА) — это гетерогенная группа наследственных заболеваний, возникающих в результате аномалий на поздних этапах эритропоэза. Такие редкие состояния характеризуются анемией разл. степени тяжести, неэффективным эритропоэзом, вторичным гемохроматозом (перегрузка железом). Они м.б. ошибочно приняты за др. виды наследственных анемий, напр. наследственный сфероцитоз/талассемия.
Дизэритропоэз является основной причиной анемии, но укороченный период полураспада циркулирующих эритроцитов также может способствовать развитию анемии. Врожденные ДЭА исторически были классифицированы на 3 основных типа (I, II и II) на основе отличительных особенностей морфологии костного мозга, клинических признаков и генетических вариантов, хотя были также определены дополнительные подгруппы и варианты (GATA1 и KLF1).
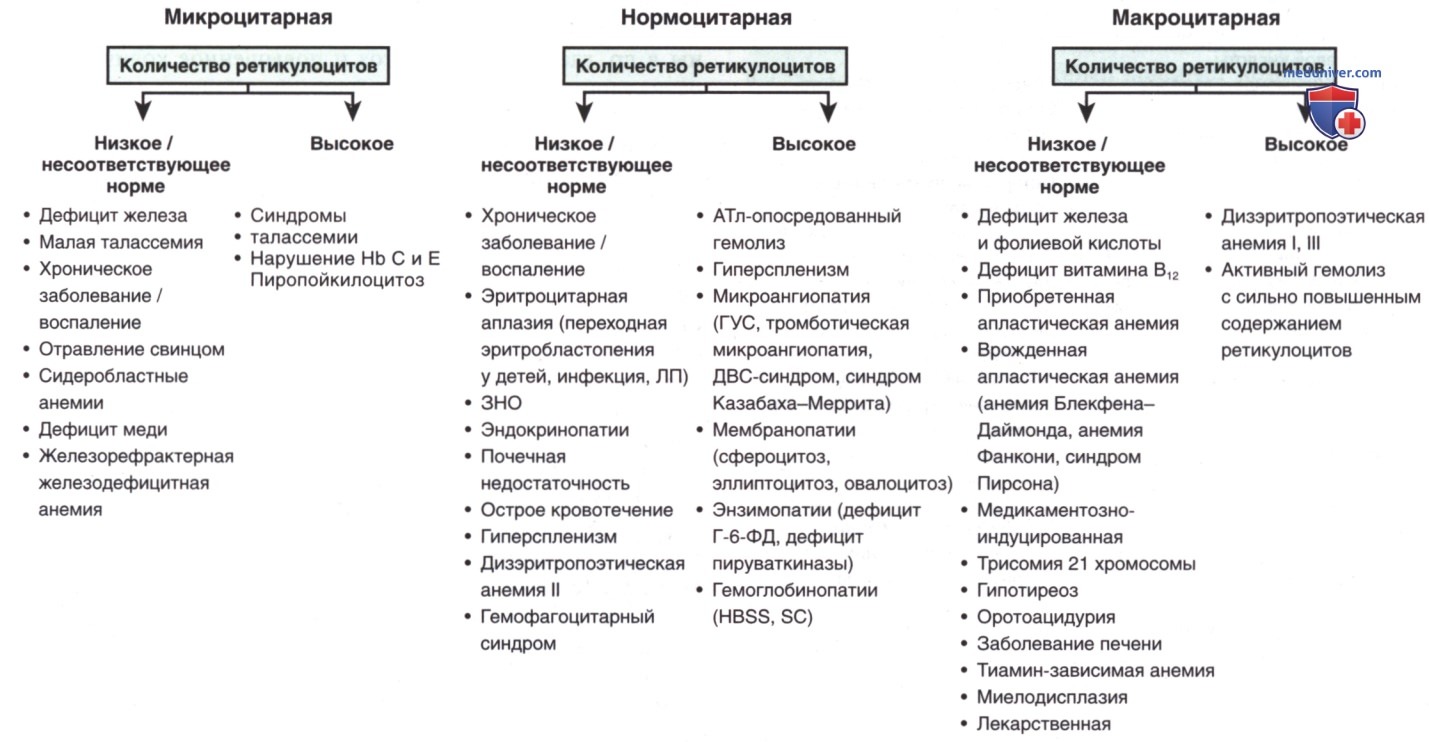
Использование показателей среднего эритроцитарного объема и подсчета ретикулоцитов при диагностике анемии
а) Врожденная дизэритропоэтическая анемия I типа:
1. Патогенез. Врожденные ДЭА I типа представляет собой АуР-расстройство. Каузальный ген (CDAN1) картирован на участке хромосомы 15 между q15.1 и q15.3. Ген кодирует коданин-1, который является повсеместно экспрессированным белком, способным ускорить сборку гистонов в хроматин и регулировать клеточный цикл. Хотя у большинства пациентов с особенностями костного мозга, свидетельствующими о наличии врожденной ДЭА I типа, есть мутации в гене CDAN1, у 11% семей такие мутации обнаружены не были. Две отличительные мутации в гене C15ORF41, который предположительно кодирует эндонуклеазу, были идентифицированы в 3 разл. генеалогических схемах врожденной ДЭА I типа.
2. Клинические проявления. Сообщается о >300 зарегистрированных случаях. Несмотря на то что врожденная ДЭА I типа м.б. диагностирована в любом возрасте, большинство случаев выявляется в детском/юношеском. Врожденная ДЭА I типа редко диагностируется в период в/утробного развития. Помимо симптомов, обусловленных анемией, к числу признаков заболевания часто относятся спленомегалия, желтуха и гепатомегалия. В более тяжелых случаях могут наблюдаться признаки экстрамедуллярного кроветворения в лобных/теменных костях черепа и в паравертебральных зонах.
Со временем развиваются холелитиаз и перегрузка железом. У 4-14% пациентов врожденная ДЭА I типа сопровождается дисморфиями, затрагивающими, в первую очередь, пальцы (синдактилия, отсутствие ногтей, полидактилия нижних кончностей). Также были зарегистрированы ангиоидные полосы сетчатки и макулярная дегенерация.
3. Результаты лабораторных исследований. Концентрация Hb обычно составляет 7-11 г/дл. Анемия обычно макроцитарная (средний эритроцитарный объем 100-120 фл), но в детском возрасте могут наблюдаться нормоцитарные показатели. Анизопойкилоцитоз определяется по мазку периферической крови. В некоторых случаях могут выявляться нормобласты и базофильная зернистость эритроцитов. Количество ретикулоцитов не соответствует степени анемии. Лабораторные данные могут свидетельствовать о перегрузке железом. В аспирате костного мозга обнаруживают эритроидную гиперплазию, мегалобластоз и базофильную зернистость. Также отмечаются двухъядерные и, реже, многоядерные полихроматофильные эритробласты.
Для врожденной ДЭА I типа высокоспецифичны не полностью разделенные клетки с тонкими хроматиновыми мостиками между ядрами пары эритроцитов. Электронная микроскопия является «золотым стандартом» диагностики, с ее помощью выявляются эритробласты с характерным паттерном гетерохроматина по типу «швейцарского сыра».
4. Лечение. Лечение в первую очередь заключается в поддерживающей терапии. Для 50% новорожденных с врожденной ДЭА I типа потребуется, по крайней мере, одна гемотрансфузия эритроцитов, а некоторые пациенты могут и в дальнейшем оставаться них зависимыми. Подросткам и взрослым гемотрансфузии могут требоваться время от времени, но только во время апластических кризов, инфекционных заболеваний и беременности.
Если анемия усугубляется сопутствующими наследственными заболеваниями, такими как талассемия/эритроцитарная энзи-мопатия, пациент может стать зависимым от данной процедуры. Лечение с помощью интерферона альфа может снизить потребность в трансфузиях. При этом пациенты не реагируют на введение эритропоэтина. Спленэктомия в большинстве случаев не рекомендуется.
Для удаления пигментных желчных камней часто требуется холецистэктомия. В нескольких тяжелых случаях была успешно проведена аллогенная трансплантация костного мозга от родного брата/сестры, совместимых по лейкоцитарному АГн человека (HLA-совместимого).
Наиболее значимым долгосрочным осложнением является гемосидероз, вызванный повышенным поглощением железа энтероцитами, неэффективным эритропоэзом и гемотрансфузионной терапией. К нормализации концентраций ферритина приводит регулярная флеботомия. Если этот метод не подходит, а уровни ферритина при повторном анализе >1,000 мкг/л/повышено содержание железа в печени, выявленное на ее R2* МРТ, следует применять пероральную хелатирующую терапию.
б) Врожденная дизэритропоэтическая анемия II типа:
1. Патогенез. Врожденная ДЭА II типа также является АуР-заболеванием. Анализ сцепления между генными локусами определил область хромосомы 20p11.2 как локализацию гена-кандидата CDAN2, который позже был идентифицирован как ген SEC23B. Известно, что этот ген кодирует цитоплазматический белок окаймления (СОР) II — компонент SEC23B, который участвует в направленной миграции везикул эндоплазматического ретикулума. Большинство случаев врожденной ДЭА II типа были обусловлены мутациями в гене SEC23B.
2. Клинические проявления. Сообщается о >450 зарегистрированных случаях, что делает врожденную ДЭА II типа наиболее распространенной формой врожденной ДЭА. Случаи врожденной ДЭА II типа в основном были зарегистрированы в странах Европы и Ближнего Востока. В отличие от врожденной ДЭА I типа, диагноз обычно ставится позже, часто потому, что симптомы м.б. более мягкими. Кроме того, врожденная ДЭА II типа м.б. первоначально ошибочно принята за наследственный сфероцитоз. Характерные признаки могут включать анемию, желтуху, спленомега-лию/гепатомегалию. Могут наблюдаться образования экстрамедуллярной гемопоэтической ткани в области заднего средостения/в паравертебральной области, возможна перегрузка железом.
3. Результаты лабораторных исследований. Нормоцитарная анемия, как правило, легкой степени с низким числом ретикулоцитов. Уровни Hb у детей ниже, чем у взрослых, и колеблются в пределах 8-11 г/дл. Наблюдается анизопойкилоцитоз, в мазке крови м.б. обнаружена нерегулярная базофильная зернистость, некоторое количество зрелых эритробластов, иногда двухъядерных. Аспират костного мозга нормобластический, но гиперклеточный, с эритроидной гиперплазией. В отличие от врожденной ДЭА I типа, наблюдается множество двухъядерных поздних полихроматических эритробластов (10-35%), а также некоторое количество многоядерных эритробластов. Могут присутствовать псевдо-клетки Гоше (Gaucher).
На электронограммах видны везикулы, нагруженные белками эндоплазматического ретикулума под клеточной мембраной. Патогномоничным признаком врожденной ДЭА II типа является лизис эритроцитов пациента в подкисленной сыворотке, вызванный IgM, который распознает АГн, присутствующий на патологических и отсутствующий на нормальных клетках.
Врожденная ДЭА II типа также известна под аббревиатурой HEMPAS (врожденная эритробластная многоядерность с «+» тестом с подкисленной сывороткой), так как она характеризуется как многоядерностью эритробластов, так и циркулирующими эритроцитами, чувствительными к лизису в подкисленной нормальной сыворотке крови. Поскольку этот тест технически сложен, диагноз обычно ставится путем анализа белков мембраны эритроцитов с помощью электрофореза в полиакриламидном геле с додецилсульфатом натрия (ДСН-ПААГ-электрофорез). При врожденной ДЭА II типа наблюдаются более тонкие полосы и более быстрая миграция переносчика анионов эритроцитов (ЕА1)/белков полосы 3 и полосы 4,5.
4. Лечение. Примерно 10% пациентов будут нуждаться в гемотрансфузии эритроцитов в младенческом и детском возрасте, но редко в зрелом возрасте. В отличие от врожденной ДЭА I типа, при врожденной ДЭА II типа спленэктомия может способствовать гематологическому улучшению. Однако она не предотвращает дальнейшую перегрузку железом даже у пациентов с нормализовавшимся уровнем Hb, предположительно из-за стойкого неэффективного эритропоэза в костном мозге. Как и в случае врожденной ДЭА I типа, вторичный гемохроматоз в данном случае является наиболее распространенным долгосрочным осложнением, и к его лечению следует подходить так, как было описано выше.
У нескольких пациентов с врожденной ДЭА II типа была успешно проведена аллогенная трансплантация костного мозга. Большинство пациентов могут вести нормальный образ жизни и иметь нормальную продолжительность жизни в случае принятия надлежащих мер в отношении предотвращения осложнений и последствий заболевания.
в) Врожденная дизэритропоэтическая анемия III типа. Врожденная ДЭА III типа — чрезвычайно редкое, мало изученное расстройство, характеризующееся легкой/умеренной макроцитарной анемией. Врожденная ДЭА III типа наследуется по АуД-типу, хотя были случаи мутаций de novol др. паттернов наследования. При врожденной ДЭА III типа мутация происходит в гене KIF23, который кодирует повсеместно распространенный белок, митотический кинезин-подобный белок 1, регулирующий разделение дочерних клеток во время митоза. В отличие от др. типов врожденной ДЭА, перегрузка железом не является клинически значимой (вероятно, потому что гемолиз преимущественно в/сосудистый), а размер селезенки обычно в норме.
У пациентов могут наблюдаться ангиоидные полосы сетчатки с макулярной дегенерацией. В мазке крови видны макроциты, анизопойкилоцитоз и нерегулярная базофильная зернистость. В костном мозге выделяются гигантские эритроидные предшественники, часто многоядерные, содержащие <12 ядер/кл. Такие многоядерные эритробласты можно увидеть и при миелодисплазии и эритролейкемии. Гемотрансфузии, как правило, не требуются.