Анемия Даймонда-Блэкфана (АДБ, Diamond, Blackfan) — редкий врожденный синдром недостаточности костного мозга, который клинически проявляется обычно в раннем младенческом возрасте. Более 90% случаев выявляется на первом году жизни. Это заболевание характеризуется анемией, как правило, нормохромной и макроцитарной; ретикулоцитопенией, недостаточным количеством/отсутствием предшественников эритроцитов в костном мозге. До 50% людей, страдающих этим заболеванием, имеют др. внегемопоэтические патологические состояния.
а) Этиология. Мутации, вызывающие АДБ, были впервые выявлены в 1997 г. в гене RPS19, который кодирует компонентный белок малой 40S-субъединицы рибосомы. Такие мутации в гене RPS19, все с АуД-типом наследования, были обнаружены у 25% пациентов. Были идентифицированы и др. гены рибосомальных белков, каждый из которых кодирует свой белок малой (40S)/большой (60S) субъединицы рибосомы. Мутации в генах рибосомальных белков были в итоге выявлены в 70% случаев, большинство из них имели АуД-наследование.
Выявление и описание новых мутаций все еще продолжаются. Поскольку большинство причинных мутаций происходит в генах рибосомальных белков, это заболевание часто называют рибосомопатией.
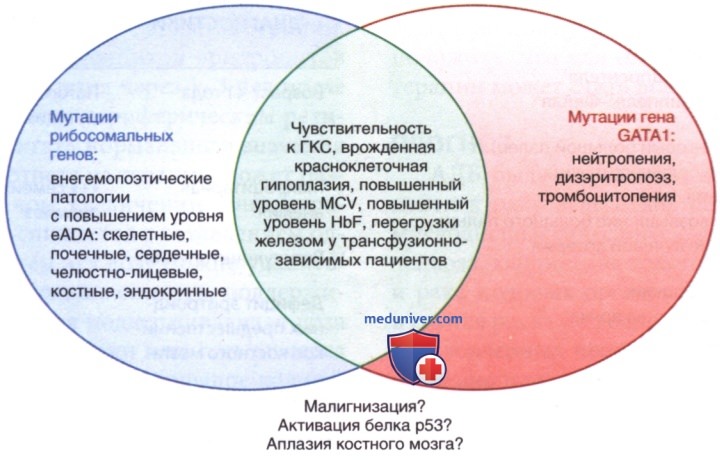
Ген GATA1, не являющийся геном рибосомальных белков, также обусловливает развитие АДБ. Мутации в гене GATA1 наследуются как рецессивные, сцепленные с Х-хромосомой, и обычно не имеют внегемопоэтических проявлений. Остается неясным, являются ли два этих пути — связанные с рибосомальной дисфункцией и нарушением производства GATA1 — независимыми друг от друга причинами одного и того же фенотипа/наоборот, АДБ является результатом нарушения единственного пути, затрагивающего функциональные связи между рибосомами и GATA1 (рис. ниже).
б) Эпидемиология. АДБ встречается у 7:1 000 000 родившихся живыми детей. Это заболевание имеет в первую очередь АуД-тип наследования, хотя могут встречаться и др. варианты. Примечательно, что существует значительное фенотипическое разнообразие АДБ даже в семьях, члены которых подвержены одной и той же мутации, что позволяет предположить влияние др. генетических модификаторов на фенотипическую экспрессию заболевания.
В рекомендациях, принятых на основе международного консенсуса, предлагается, чтобы диагноз «неклассическая картина» АДБ применялся к членам семьи, у которых выявлена мутация/к членам семьи, у которых мутация не выявлена, но имеется сопутствующая патология/отклонения лабораторных показателей от нормы.
в) Клинические проявления. Выраженная анемия обычно проявляется ко второму месяцу жизни, иногда несколько позже. При рождении анемия отмечается у 25% пациентов, при этом отечный синдром новорожденных встречается редко; у 92% заболевание диагностируется в первый год жизни. Примерно 40-50% пациентов имеют ВПР, а у 25% пациентов с АДБ обнаружено >1 патологии (табл. 1). Наиболее распространенными (у 50% пациентов) являются аномалии черепно-лицевой области, которые включают в себя короткий и плоский нос, готическое нёбо.
Аномалии скелета, в основном верхних конечностей и кистей рук, обнаружены у 30%. Пороки развития большого пальца кисти, включая плоское возвышение и трехфаланговость большого пальца, м.б. двусторонними/односторонними. Может отсутствовать лучевая артерия. Были выявлены пороки развития МПС (38%) и ССС (30%), а также офтальмологические и костно-мышечные нарушения. Часто встречается низкий рост, но обычно неясно, является ли эта характеристика результатом самого заболевания/связанных с ним методов лечения/совокупности этих причин.
г) Результаты лабораторных исследований. Эритроциты обычно макроцитарны для возраста, но гиперсегментированные нейтрофилы/др. признаки мегалобластной анемии в мазке периферической крови не обнаруживаются. Ферментные паттерны эритроцитов схожи с паттернами «фетальной» популяции эритроцитов, с повышенной экспрессией АГн "i" и повышенным фетальным Hb (HbF). У большинства пациентов с АДБ активность эритроцитарной аденозиндезаминазы увеличена, что помогает отличить врожденную красноклеточную аплазию от приобретенной ТЭД.
Поскольку при нормальных фетальных эритроцитах не происходит повышения активности эритроцитарной аденозиндезаминазы, измерение этого фермента м.б. особенно полезным при диагностике АДБ у детей первых месяцев жизни. Также могут наблюдаться тромбоцитоз/ред-ко тромбоцитопения, а иногда нейтропения. Содержание ретикулоцитов резко снижено, несмотря на анемию тяжелой степени. Количество предшественников эритроцитов костного мозга у большинства пациентов значительно снижено; др. элементы костного мозга, как правило, находятся в норме.
Уровень сывороточного железа повышен. В отличие от анемии Фанкони (Fanconi), при воздействии алкилирующих агентов на лимфоциты не наблюдается повышения количества хромосомных разрывов. В табл. 2 представлены рекомендуемые критерии диагностики.
д) Дифференциальная диагностика. АДБ следует дифференцировать от др. видов анемий, сопровождающихся низким содержанием ретикулоцитов. Синдром ТЭД часто является основным альтернативным диагнозом. В табл. 1 представлено практическое сравнение данных по этим двум заболеваниям. ТЭД часто отличается от АДБ относительно поздним началом, хотя изредка развивается и у младенцев <6 мес. Макроцитоз, ВПР, признаки фетальных эритроцитов и повышенный уровень эритроцитарной аденозиндезаминазы обычно являются признаками АДБ, а не ТЭД.
Следует также рассмотреть др. наследственные синдромы макроцитарной недостаточности костного мозга, в частности, анемию Фанкони и синдром Швахмана-Даймонда (Shwachman), миелодиспластический синдром. Синдром Аазе (Aase) включает врожденную красноклеточную аплазию с трехфаланговым большим пальцем, ВПС и расщелину нёба. Гемолитическая болезнь новорожденных также может имитировать характерные признаки АДБ, так как может иметь затяжное течение и сопровождаться значительным снижением эритропоэза. Анемия при этом заболевании обычно купируется самостоятельно через 5-8 нед.
Тяжело протекающие хронические гемолитические заболевания могут осложняться апластическим кризом, характеризующимся ретикулоцитопенией и снижением количества предшественников эритроцитов в костном мозге. Такое явление обычно наблюдается после нескольких месяцев жизни и часто вызывается парвовирусной В 19-инфекцией. В/утробная парвовирусная В19-инфекция может проявляться изолированной красноклеточной аплазией в младенчестве и даже отечным синдромом новорожденных. При диагностике АДБ у младенцев первых месяцев жизни важно исключить парвовирусную В19-инфекцию методом ПЦР.
Должны быть исключены др. инфекции, включая ВИЧ, воздействие ЛП, иммунные процессы и синдром Пирсона (Pearson).
е) Лечение. Главным направлением в терапии АДБ является применение ГКС, и у ~80% пациентов имеется первоначальный ответ на терапию. Поскольку ГКС тормозят линейный рост, физическое и нейрокогнитивное развитие, многие гематологи используют в лечении длительную гемотрансфузионную терапию, откладывая начало приема ГКС до 1 года. Преднизон/преднизолон, 2 мг/кг в сутки, применяются в качестве пробного варианта терапии.
Увеличение количества предшественников эритроцитов в костном мозге обычно наблюдается через 1-3 нед после начала терапии и сопровождается периферическим ретикулоцитозом. Hb может достигать нормального значения через 4-6 нед, хотя скорость ответа на терапию может различаться. При подтвержденном увеличении концентрации Hb доза ГКС постепенно снижается с оставлением одной, эффективной минСД. Затем эту дозу можно удвоить/использовать через день/еще больше снизить, поддерживая Hb на уровне >9 г/дл. Целевая поддерживающая доза не должна превышать 0,5 мг/кг в сутки или 1 мг/кг через день.
Для некоторых пациентов очень небольшое количество преднизона, всего 2,5 мг 2 р/нед, м.б. достаточным для поддержания нормального эритропоэза. Плановые контрольные обследования и тестирования на побочные эффекты ГКС должны проводиться у всех пациентов, независимо от дозы. Надлежащая профилактика пневмоцистной пневмонии должна проводиться после 1 мес приема высоких доз ГКС и продолжаться до тех пор, пока не будут достигнуты низкие дозы ЛП с приемом через день. Многие дети с АДБ обычно перестают принимать ГКС вследствие нежелательных побочных эффектов/при развитии рефрактерности к данным ЛП.
Длительные гемотрансфузии эритроцитов необходимы 35% пациентов, включая пациентов с отсутствием ответа на ГКС (30%), стероид-рефрактерных пациентов (15%) и пациентов, которые не м.б. отлучены от груди для получения приемлемой низкой дозы (50%). Для поддержания уровня Hb >8 г/дл, гемотрансфузии проводятся с интервалом в 3-5 нед. Некоторым детям младшего возраста для обеспечения нормального развития и жизнедеятельности может потребоваться уровень Hb >9 г/дл.
При перегрузке железом у трансфузионно-зависимых пациентов необходим соответствующий скрининг и, в перспективе, введение хелатирующей терапии. В одном случае пациент с АДБ, получавший L-лейцин, перестал зависеть от гемотрансфузии и оставался в ремиссии >5 мес. В настоящее время ведутся дальнейшие доклинические и клинические исследования.
Были получены данные о спонтанной ремиссии анемии независимо от ГКС-терапии/гемотрансфузии эритроцитов. Вероятность ремиссии составляет 25% к возрасту 25 лет, причем у большинства пациентов ремиссия наблюдается в течение первого десятилетия. При этих условиях сохраняется легкая макроцитарная анемия и повышенный уровень эритроцитарной аденозиндезаминазы.
М.б. эффективной ТГСК. Лучшие результаты у пациентов <9 лет достигаются при подборе доноров из числа родных братьев/сестер, совместимых по системе лейкоцитарных АГн человека (HLA-система). ТГСК от HLA-совместимого родного брата/сестры рекомендуется детям раннего возраста, зависимых от гемотрансфузии. Некоторые рекомендуют проводить трансплантацию в 3-9 лет, другие же настаивают на проведении данного вида терапии в более молодом возрасте с целью предотвращения перегрузки железом и аллогенной сенсибилизации при длительных гемотрансфузиях эритроцитов.
Важно тщательно обследовать родственных доноров, включая анализ генотипа, если он известен, чтобы удостовериться, что донор не является носителем гена АДБ, как сам пациент. Усовершенствования метода альтернативного лечения посредством ТГСК от донора позволяют предположить, что для определенных пациентов данный вид терапии может стать востребованным.
ж) Прогноз. АДБ была определена как синдром предрасположенности к раку вследствие более высокого риска развития миелодиспластического синдрома, острого миелоидного лейкоза, карциномы толстой кишки, остеогенной саркомы и рака половых органов у женщин. Пациенты находятся в группе риска по возникновению эндокринных патологий, обусловленных перегрузкой железом (СД, гипогонадизм), особенно при гемотрасфузиях.
Пациенты, прошедшие процедуру ТГСК, подвержены риску отдаленных последствий, обусловленных этой процедурой. Общая актуарная выживаемость всех пациентов с АДБ в возрасте 40 лет составляет 75%, из них ок. 87% пациентов получают ГКС и ок. 57% зависят от гемотрансфузии. Из зарегистрированных смертей 67% были связаны с лечением и 22% с АДБ (ЗНО и тяжелая апластическая анемия).
Данные о результатах лечения и выживаемости собираются с помощью Реестра анемии Даймонда-Блэкфана.