Синдром Альпорта, или наследственный нефрит, — это генетически гетерогенное заболевание, вызванное мутациями в генах, кодирующих коллаген IV типа, основной компонент базальных мембран. Эти генетические изменения определяют вариабельность клинической картины, особенности течения болезни и гистологические повреждения.
а) Генетика. У ~85% пациентов имеется Х-сцепленное наследование, вызванное мутацией в гене COL4A5, кодирующем α5-цепь коллагена IV типа. Пациенты с подтипом Х-сцепленного синдрома Альпорта и диффузным лейомиоматозом демонстрируют непрерывную мутацию в генах COL4A5 и COL4A6, кодирующих α5- и α6-цепи коллагена IV типа, соответственно. Аутосомно-рецессивные формы синдрома Альпорта у 15% пациентов вызываются мутациями в генах COL4A3 и COL4A4 на хромосоме 2, кодирующих α3- и α4-цепи коллагена IV типа, соответственно. АуД-доминантная форма синдрома Альпорта, сцепленная с локусом гена COL4A3-COL4A4, встречается в 5%.
Синдром Фехтнера (синдром Альпорта с макротромбоцитопенией) — АуД-заболевание, вызванное мутациями в MYH9. Наследственная ангиопатия с нефропатией, аневризмой и мышечными спазмами может изначально напоминать синдром Альпорта. Она возникает из-за мутаций в гене COL4A.
б) Патология. Образцы биопсии почек в течение первого десятилетия жизни покажут лишь несколько изменений при световой микроскопии. Позже в клубочках могут развиться мезангиальная пролиферация и утолщение стенки капилляров, приводя к прогрессирующему склерозу клубочков. По мере прогрессирования заболевания развиваются атрофия канальцев, интерстициальное воспаление и фиброз, а также содержащие липиды трубчатые или интерстициальные клетки, называемые «пенистыми» клетками. Иммунопатологические исследования обычно не являются диагностическими.
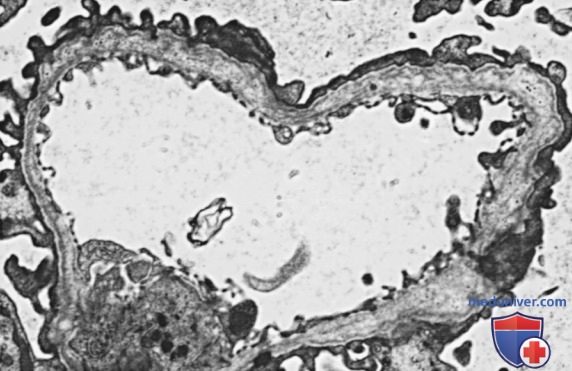
У большинства пациентов электронная микроскопия выявляет диффузное утолщение, истончение, расщепление и расслоение клубочковой и канальцевой базальных мембран (рис. ниже). Ультраструктурный анализ гломерулярной базальной мембраны при всех генетических формах синдрома Альпорта м.б. полностью нормальным, отображать неспецифические изменения или демонстрировать только равномерное истончение, что усложняет постановку диагноза.
Электронная микрофотография биоптата ребенка с синдромом Альпорта, показывающая утолщение, истончение, расщепление и расслоение гломерулярной базальной мембраны (х 1650)
в) Клинические проявления. У всех пациентов с синдромом Альпорта наблюдается бессимптомная микрогематурия, которая м.б. интермиттирующей у женщин и более молодых мужчин. Единичные или повторяющиеся эпизоды макрогематурии, обычно возникающие через 1-2 дня после инфекции ВДП, наблюдаются у ~50% пациентов. Протеинурия часто наблюдается у мужчин, но может отсутствовать, быть легкой или интермиттирующей у женщин.
Прогрессирующая протеинурия, часто >1 г/сут, диагностируется во втором десятилетии жизни и впоследствии может привести к развитии нефротического синдрома.
Двусторонняя нейросенсорная тугоухость, которая никогда не бывает врожденной, развивается у 90% гемизиготных мужчин с Х-сцепленным синдромом Альпорта, у 10% гетерозиготных женщин с Х-сцепленным синдромом Альпорта и у 67% пациентов с аутосомно-рецессивным синдромом Альпорта. Этот дефицит начинается в диапазоне высоких частот, но прогрессирует, затрагивая слух, связанный с нормальной речью, что вызывает необходимость в слуховых аппаратах.
Такое прогрессирование потери слуха идет параллельно с потерей функции почек. Глазные аномалии, которые встречаются у 30-40% пациентов с Х-сцепленным синдромом Альпорта, включают передний лентиконус (вытеснение центральной части хрусталика в переднюю камеру), макулярные пятна и эрозии роговицы. Есть сообщения о лейомиоматозе пищевода, трахеобронхиального дерева и женских половых органов, но это встречается редко.
г) Диагностика. Сочетание положительного семейного анамнеза, скринингового ОАМ родственников первой степени родства, аудиограммы и офтальмологического обследования имеет решающее значение для постановки диагноза синдрома Альпорта. Наличие переднего лентиконуса патогномонично.
Синдром Альпорта вероятен у пациента с гематурией и, по крайней мере, двумя из следующих характерных клинических признаков: макулярные пятна, рецидивирующие эрозии роговицы, утолщение и истончение гломерулярной базальной мембраны или нейросенсорная глухота. Отсутствие окрашивания базальной мембраны эпидермиса на α5-цепь коллагена IV типа у гемизигот мужского пола и прерывистое окрашивание базальной мембраны эпидермиса у гетерозигот женского пола при биопсии кожи являются патогномоничными для Х-сцепленного синдрома Альпорта и м.б. противопоказанием для диагностической биопсии почек.
Клинически показано генетическое тестирование на наличие мутаций Х-сцепленного синдрома Альпорта и COL4A5. Пренатальная диагностика необходима для семей, в которых есть члены с Х-сцепленным синдромом Альпорта и носители идентифицированной мутации.
д) Прогноз и лечение. Риск прогрессирующей почечной дисфункции, ведущей к терминальной стадии почечной недостаточности, наиболее высок среди гемизигот и АуР-гомозигот. Терминальная стадия почечной недостаточности возникает в возрасте до 30 лет у 75% гемизигот с Х-сцепленным синдрома Альпорта. Риск терминальной стадии почечной недостаточности у Х-сцепленных гетерозигот составляет 12% к 40 годам и 30% к 60 годам.
Факторами риска прогрессирования являются макрогематурия в детстве, нефротический синдром и заметное утолщение гломерулярной базальной мембраны. Внутрисемейные вариации фенотипической экспрессии приводят к значимым различиям в возрасте развития терминальной стадии почечной недостаточности среди членов семьи. Специфической терапии для лечения синдрома Альпорта не существует, хотя иАПФ (и, возможно, ингибиторы рецепторов ангиотензина-2) могут замедлять скорость прогрессирования. Правильное лечение осложнений почечной недостаточности, таких как АГ, анемия и нарушение электролитного баланса, имеет решающее значение.
Пациентам с терминальной стадией почечной недостаточности проводится диализ и трансплантация почки. Приблизительно у 5% реципиентов в трансплантированной почке развивается антигломерулярно-базально-мембранный нефрит, возникающий в основном у мужчин в возрасте до 30 лет с Х-сцепленным синдромом Альпорта и терминальной стадией почечной недостаточности.
Фармакологическое лечение протеинурии иАПФ или блокаторами рецептора ангиотензина-2 доказало свою эффективность при других гломерулярных заболеваниях, а также представляет многообещающие результаты при синдроме Альпорта. Также рекомендуется скрининг гетерозиготных носителей при тяжелых заболеваниях почек в более зрелом возрасте и возможным лечением выраженной протеинурии.