Болезнь Гудпасчера — это аутоиммунное заболевание, характеризующееся легочным кровотечением, быстропрогрессирующим гломерулонефритом и повышенными титрами АТл к гломерулярной базальной мембране (ГБМ).
Заболевание возникает в результате аутоиммунного ответа, направленного против определенных эпитопов коллагена IV типа, расположенных в базальной альвеолярной мембране легких и гломерулярной базальной мембране почек. Приобретенное конформационное изменение неколлагенового домена-1 α3-цепи коллагена IV типа приводит к продукции патологических ауто-АТл.
Высокое сродство этих АТл к ГБМ приводит к характерному быстропрогрессирующему заболеванию почек. Введение человеческих АТл к ГБМ животным воспроизводит быстропрогрессирующий гломерулонефрит, подтверждая высокую патогенность этих АТл.
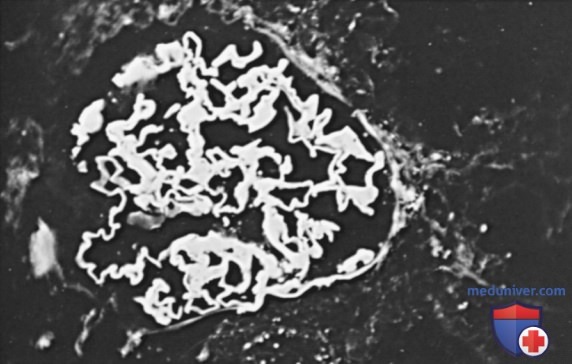
а) Патологическая анатомия. При биопсии почек у большинства пациентов выявляется полулунный гломерулонефрит. При иммунофлуоресцентной микроскопия определяется патогномоничные непрерывные линейные депозиты IgG вдоль ГБМ (см. рис. ниже).
Иммунофлуоресцентная микрофотография, демонстрирующая непрерывное линейное окрашивание IgG на протяжении гломерулярной базальной мембраны при болезни Гудпасчера (х250)
б) Клинические проявления. Болезнь Гудпасчера у детей встречается редко. У пациентов обычно наблюдается кровохарканье по причине легочного кровотечения, которое м.б. жизнеугрожающим. Сопутствующие почечные проявления включают в себя острый гломерулонефрит с гематурией, нефритический мочевой осадок с клеточными цилиндрами, протеинурию и АГ, которые обычно быстро прогрессируют. ХПН обычно развивается в течение нескольких дней или недель после начала клинических проявлений.
Может присутствовать лихорадка, но др. системные жалобы, такие как общая слабость или артралгия, обычно отсутствуют; их наличие должно вызвать настороженность относительно наличия системного васкулита. Реже пациенты могут иметь анти-ГБМ нефрит, проявляющийся в виде изолированного, быстропрогрессирующего гломерулонефрита без легочного кровотечения. Практически во всех случаях анти-ГБМ АТл присутствуют в плазме крови и/или паренхиме почек, и уровень С3 сывороточного комплемента является нормальным.
Уровень антинейтрофильных цитоплазматических АТл м.б. повышен у 10-40% пациентов, наряду с анти-ГБМ АТл; у пациентов с двойным положительным результатом на вышеуказанные ауто-АТл при обращении наблюдается более тяжелое течение процесса. Как правило, титр АТл к ГБМ коррелируют с тяжестью поражения почек. Однако следует выполнить биопсию почки (если нет противопоказаний), поскольку точность серологических исследований на анти-ГБМ АТл варьирует, а биопсия почек дает дополнительную информацию, которая м.б. информативной для определения тактики лечения.
в) Диагноз и дифференциальная диагностика. Диагноз устанавливается на основании данных клинической картины легочного кровотечения в сочетании с острым гломерулонефритом; наличия сывороточных АТл к ГБМ (к коллагену IV типа в ГБМ); и характерных результатов биопсии почек.
Необходимо учитывать и др. заболевания, которые могут вызывать легочно-почечный синдром, включая СКВ; ПШГ; ТЭЛА, ассоциированную с нефротическим синдромом; и васкулит, ассоциированный с антинейтрофильными цитоплазматическими АТл (напр., гранулематоз с полиангиитом и микроскопический полиангиит). Эти заболевания м.б. исключены вследствие отсутствия др. характерных клинических признаков, результатов биопсии почек и отрицательных серологических исследований на антинуклеарные АТл, АТл к двухцепочечной ДНК и антинейтрофильные цитоплазматические АТл.
г) Прогноз и лечение. Без лечения прогноз при болезни Гудпасчера плохой. Оно должно быть начато в срочном порядке при подозрении на наличие данной патологии. Своевременное начало плазмафереза, пульс-терапии метилпреднизолоном и циклофосфамидом в/в часто индуцирует ремиссию и увеличивает продолжительность жизни.
Проведение плазмафереза позволяет удалить циркулирующие анти-ГБМ АТл, а иммуносупрессивная терапии ГКС и циклофосфамидом ингибирует продукцию АТл. Ритуксимаб м.б. использован в качестве альтернативной терапии в случаях избыточной токсичности циклофосфамида. Инициальное лечение определяется клиническим ответом и титрами АТл к ГБМ.
Данные ретроспективных когортных исследований показывают, что, при раннем начале комбинированной терапии у большинства пациентов отмечается благоприятный исход для почечной функции. Однако инициальная манифестация заболевания с олигоанурического синдрома, наличия высокой доли гломерулярных полулунных очагов или ОПН, требующей диализа, является предиктором плохих исходов для почечной функции и общей выживаемости. После достижения ремиссии поддерживающая терапия более низкими дозами (преднизон + азатиоприн/микофенолата мофетил) продолжается в течение 6-9 мес.
Однако у пациентов, имеющих в анамнезе острое легочное кровотечение и быстропрогрессирующий гломерулонефрит, может развиться терминальная ХПН, несмотря на продолжающуюся иммуносупрессивную терапию. В таком случае трансплантация почки является методом выбора. Обострение и рецидивы заболевания после трансплантации почки встречаются редко.