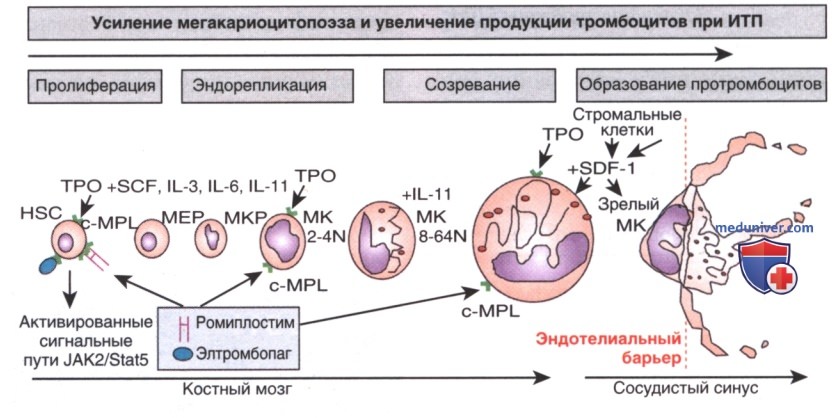
а) Мегакариоцитопоэз. Тромбоциты — это безъядерные клеточные фрагменты, продуцируемые мегакариоцитами (крупными полиплоидными клетками) в костном мозге и др. тканях. По мере созревания мегакариоцитов происходит расширение цитоплазмы и высвобождается большое количество тромбоцитов. Продолжительность жизни циркулирующих тромбоцитов составляет 10-14 дней. Тромбопоэтин является основным фактором роста, контролирующим выработку тромбоцитов (рис. 1). Уровни тромбопоэтина, по-видимому, обратно коррелируют с количеством тромбоцитов и массой мегакариоцитов.
Рисунок 1. Схема мегакариоцитопоэза и продукции тромбоцитов при идиопатической тромбоцитопенической пурпуре. Гемопоэтические стволовые клетки (HSC) мобилизуются, а мегакариоциты (МК) и эритроидные предшественники (МЕР) концентрируются с МК-коммитированными предшественниками (МКР), порождая зрелые мегакариоциты под контролем тромбопоэтина (ТРО), действующего совместно с хемокинами, цитокинами и факторами роста, включая фактор стволовых клеток (SCF) и IL: IL-3, IL-6 и IL-11. Эндорепликация приводит к изменению плоидности мегакариоцитов и увеличению числа хромосом (до 64N). Зрелые мегакариоциты мигрируют к эндотелиальному барьеру, ограничивающему сосудистый синус, и под влиянием стромального клеточного фактора-1 (SDF-1) продуцируют протромбоциты, которые выступают в кровоток и продуцируют большое количество тромбоцитов под влиянием гемодинамических детерминантных факторов. Назначенные лекарственные препараты ромиплостим и элтромбопаг проникают в костный мозг и соединяются с тромбопоэтином для стимуляции мегакариоцитопоэза и выработки тромбоцитов. c-MPL — рецептор тромбопоэтина
Уровни тромбопоэтина наиболее высоки при тромбоцитопенических состояниях, связанных со сниженным мегакариопоэзом костного мозга, и могут варьироваться при повышенном производстве тромбоцитов.
Тромбоциты играют множество ролей в системе гемостаза. На поверхности тромбоцитов располагается ряд важных рецепторов для адгезивных белков, включая фактор Виллебранда (ФВл) и фибриноген, а также там находятся рецепторы для агонистов, запускающих агрегацию тромбоцитов, такие как тромбин, коллаген и АДФ. После повреждения стенки кровеносного сосуда экспонируется внеклеточный матрикс, содержащий адгезивные и прокоагулянтные белки.
Субэндотелиальный коллаген связывает ФВл, который изменяет свою конформацию, что индуцирует связывание тромбоцитарного комплекса гликопротеина Ib (GPIb), рецептора ФВл. Этот процесс называется адгезией тромбоцитов. Затем происходит активация тромбоцитов. В процессе активации тромбоциты вырабатывают тромбоксан А2 из арахидоновой кислоты с помощью фермента ЦОГ. После активации в окружающую кровь и лимфу тромбоцитами высвобождаются агонисты, такие как АДФ, АТФ, ионы кальция, серотонин и факторы свертывания. Связывание ФВл с комплексом GPIb запускает сложный сигнальный каскад, который приводит к активации рецептора фибриногена, основного тромбоцитарного гликопротеина-интегрина aIIb-β, (GPIIb-IIIa).
Циркулирующий фибриноген связывается со своим рецептором на активированных тромбоцитах, соединяя тромбоциты между собой в процессе, называемом агрегацией тромбоцитов. В результате в месте повреждения сосудов образуется гемостатическая пробка.
Высвобождающиеся при активации серотонин и гистамин усиливают локальную вазоконстрикцию. Кроме взаимодействия со стенкой сосуда для образования тромбоцитарной пробки, тромбоцит обеспечивает каталитическую поверхность, на которой концентрируются факторы свертывания, в конечном итоге генерирующие тромбин посредством последовательной серии расщепления ферментов. Наконец, под действием сократительных белков тромбоцитов и сокращения цитоскелета происходит ретракция сгустка.
б) Тромбоцитопения. Нормальное количество тромбоцитов составляет 150— 450x109/л. Тромбоцитопения характеризуется уменьшением количества тромбоцитов до уровня <150х109/л, хотя клинически значимое кровотечение не наблюдается до тех пор, пока количество тромбоцитов не упадет значительно ниже уровня 50х109/л. Тромбоцитопении м.б. вызваны разными причинами, включая врожденные или приобретенные нарушения, связанные со снижением продукции тромбоцитов, секвестрацией тромбоцитов в увеличенной селезенке или др. органе и усиленным разрушением нормально синтезированных тромбоцитов иммунного или неиммунного характера (табл. 1 и 2, рис. 2).
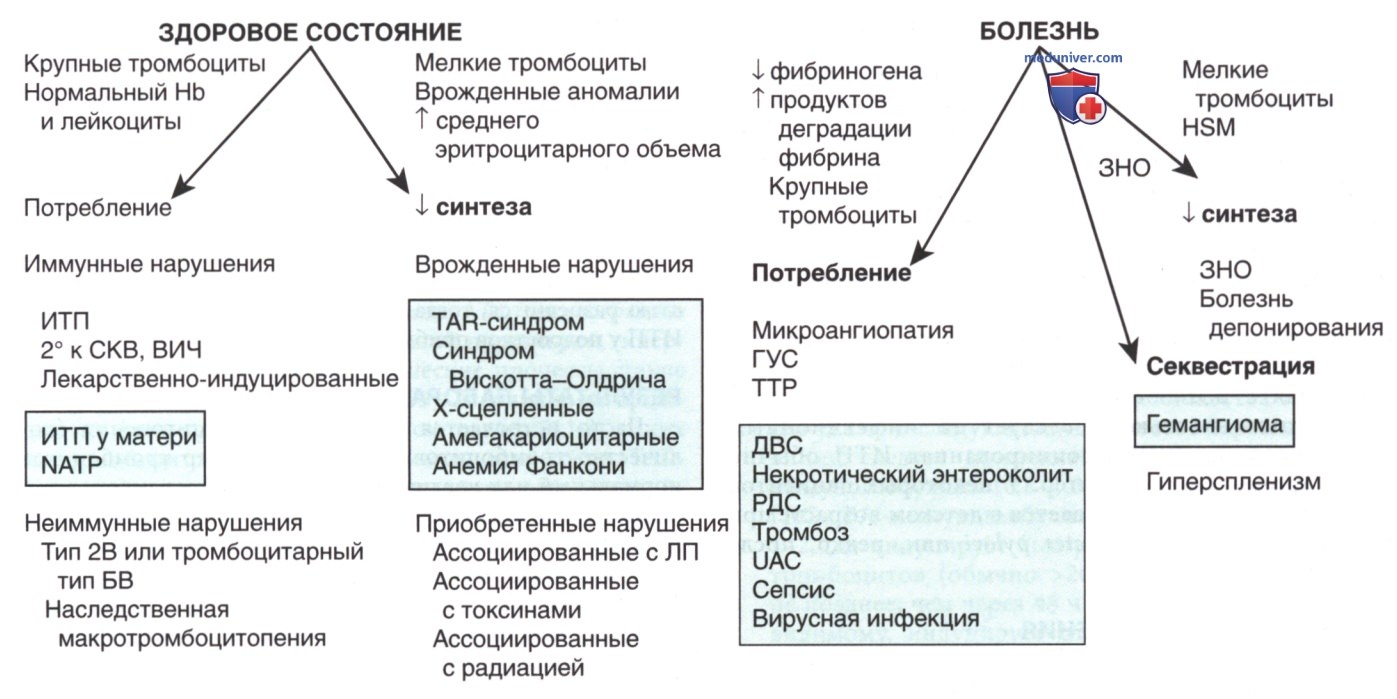
Рисунок 2. Дифференциальная диагностика тромбоцитопенических синдромов у детей. В первую очередь синдромы различаются по клинической картине. Параметры, указывающие на диагноз, выделены курсивом. Механизмы и общие нарушения, которые ведут к данным результатам, показаны в нижней части рисунка. Расстройства, которые обычно поражают новорожденных, перечислены в заштрихованных прямоугольниках. БВ — болезнь Виллебранда; WBC — лейкоциты; HSM — гепатоспленомегалия; NATP — неонатальная аллоиммунная тромбоцитопеническая пурпура; TAR-синдром (Trombocytopenia — Absent Radius syndrome) — тромбоцитопения с отсутствием лучевой кости ; ТТР — тромботическая тромбоцитопеническая пурпура; UAC — катетер пупочной артерии
в) Идиопатическая (аутоиммунная) тромбоцитопеническая пурпура. Наиболее частой причиной острого начала тромбоцитопении у здоровых в остальном детей является (аутоиммунная) ИТП.
1. Эпидемиология. У небольшого числа детей, 1:20 000, через 1-4 нед после воздействия общей вирусной инфекции развиваются аутоАТл к АГн, расположенным на поверхности тромбоцитов, что приводит к внезапному развитию тромбоцитопении. Недавний анамнез вирусного заболевания описан у 50-65% детей с ИТП. Возраст пика заболеваемости составляет 1-4 года, хотя в целом возрастной диапазон охватывает период от раннего младенчества до пожилого возраста. В детском возрасте пациенты мужского и женского пола подвержены заболеваемости одинаково. ИТП, по-видимому, чаще развивается в конце зимы и весной после пикового сезона вирусных респираторных заболеваний.
2. Патогенез. Точная антигенная мишень для большинства таких АТл при острой форме ИТП у детей чаще всего остается неопределенной, хотя при хронической форме ИТП у многих пациентов обнаруживаются АТл к аIIb-β3 и GPIb. После связывания АТл с поверхностью тромбоцита циркулирующие тромбоциты, нагруженные АТл, распознаются рецепторами к Fc-фрагменту IgE, находящимися на макрофагах селезенки, поглощаются и разрушаются. Многие распространенные вирусы были описаны как ассоциированные с ИТП, включая ВЭБ (ВЭБ) и ВИЧ. ИТП, связанная с ВЭБ, обычно характеризуется короткой продолжительностью и следует за инфекционным мононуклеозом. ВИЧ-ассоциированная ИТП обычно носит хронический характер.
У некоторых пациентов ИТП, по-видимому, развивается в детском возрасте при инфицировании Helicobacter pylori или, редко, после вакцинации.
3. Клинические проявления. Классическим проявлением ИТП является ранее здоровый ребенок 1-4 лет с внезапным развитием генерализованных петехий и пурпуры. Родители часто заявляют, что день назад ребенок был в порядке, а в настоящий момент весь покрыт синяками и фиолетовыми пятнами. Возможны кровотечения из десен и слизистых оболочек, особенно при глубокой тромбоцитопении (количество тромбоцитов <10х109/л). В анамнезе отмечается предшествующая вирусная инфекция за 1-4 нед до начала тромбоцитопении. Результаты физикального обследования нормальные, за исключением петехий и пурпуры. Спленомегалия, лимфаденопатия, боль в костях и бледность встречаются редко.
В Великобритании была предложена простая система классификации для характеристики степени тяжести кровотечения при ИТП на основе симптомов и признаков, а не на основе количества тромбоцитов, эта классификация представлена ниже:
1) Отсутствие симптомов.
2) Легкие симптомы: синяки и петехии, иногда незначительные носовые кровотечения, слабо влияющие на повседневную жизнь.
3) Умеренные симптомы: более тяжелые поражения кожи и слизистых оболочек, более обильные носовые кровотечения и меноррагии.
4) Тяжелые симптомы: эпизоды кровотечения, меноррагии, носовые кровотечения, ЖКК, требующие гемотрансфузии или госпитализации; симптомы, серьезно влияющие на качество жизни.
Наличие отклонений от нормы при обследовании, таких как гепатоспленомегалия, боль в костях или суставах, выраженная лимфаденопатия, др. цитопении или врожденные аномалии, может указывать на др. диагнозы (лейкоз, синдромы). При бессимптомном начале заболевания, особенно в подростковом возрасте, более вероятно развитие хронической формы ИТП или системного заболевания, такого как СКВ.
4. Исход. Тяжелое кровотечение встречается редко (<3% случаев в 1 крупном международном исследовании). У 70-80% детей с острой формой ИТП спонтанное разрешение заболевания происходит в течение 6 мес. Терапия, по-видимому, не влияет на естественное течение болезни. У <1% пациентов развивается в/черепное кровоизлияние. Сторонники интервенционной терапии утверждают, что целью ранней терапии является повышение количества тромбоцитов до уровня >20х109/л и предотвращение развития редко встречающегося в/черепного кровоизлияние. Доказательств того, что терапия предотвращает серьезные кровотечения, нет.
У 20% детей с острой формой ИТП развивается хроническая ИТП. Исход/прогноз м.б. в большей степени связан с возрастом; ИТП у детей более младшего возраста с большей вероятностью разрешится, тогда как развитие хронической формы ИТП у подростков приближается к 50%.
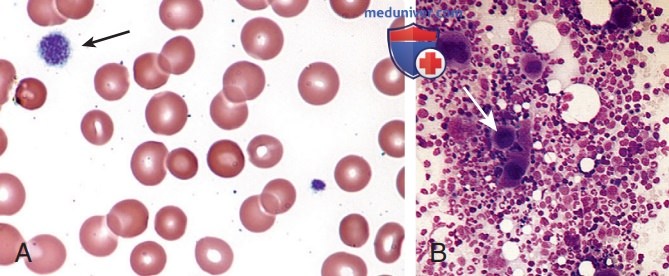
5. Результаты лабораторных исследований. Часто встречается тяжелая тромбоцитопения (количество тромбоцитов <20х109/л), размер тромбоцитов нормальный или увеличенный, что отражает ускоренный оборот тромбоцитов (рис. 3). При острой форме ИТП показатели Hb, количество лейкоцитов и определение лейкоцитарной формулы должны быть в норме. Уровень Hb м.б. снижен при обильном носовом кровотечении (эпистаксисе) или меноррагии. Исследование костного мозга показывает нормальные клетки гранулоцитарного и эритроцитарного ряда с характерно нормальным или повышенным количеством мегакариоцитов.
Рисунок 3. Аспират крови и костного мозга ребенка с идиопатической тромбоцитопенической пурпурой: А — в мазке крови видны крупные тромбоциты; В — аспират костного мозга показывает повышенное количество мегакариоцитов, многие из которых кажутся незрелыми
Некоторые из мегакариоцитов могут казаться незрелыми, что отражает ускоренный оборот тромбоцитов. Показанием к аспира-ции/биопсии костного мозга является аномальное количество лейкоцитов, или дифференцированная либо необъяснимая анемия, а также данные анамнеза и физикального обследования, указывающие на синдром недостаточности костного мозга или ЗНО. Др. лабораторные исследования должны проводиться на основании анамнеза и физикального обследования. Исследования на ВИЧ следует проводить в группах риска, особенно среди сексуально активных подростков. Тест на АТл к тромбоцитам редко бывает полезен при острой форме ИТП.
Прямой антиглобулиновый тест (прямая реакция Кумбса (Coombs, R.R.A.)) следует проводить при наличии необъяснимой анемии, чтобы исключить синдром Эванса (Evans, R.S.) (аутоиммунную гемолитическую анемию и тромбоцитопению). Синдром Эванса м.б. идиопатическим или ранним признаком СКВ, аутоиммунного лимфопролиферативного синдрома или синдрома вариабельного неклассифицируемого иммунодефицита. Следует рассматривать необходимость анализа на уровень антинуклеарного фактора у подростков, особенно если у них обнаруживаются др. признаки СКВ.
6. Диагностика и дифференциальная диагностика. Выглядящий здоровым ребенок с тромбоцитопенией умеренной или тяжелой степени, нормальными результатами ОАК и нормальными результатами физикального обследования должен пройти ДД на ограниченное число нарушений, которые включают воздействие ЛС, индуцирующих лекарственно-зависимые АТл, секвестрацию селезенки из-за ранее недооцененной портальной гипертензии и, редко, ранние апластические процессы, такие как анемия Фанкони.
В отличие от врожденных форм тромбоцитопении, таких как тромбоцитопения с отсутствием лучевой кости (TAR-синдром) и тромбоцитопения, ассоциированная с мутацией в гене MYH9, др. синдромы, включающие большинство процессов, происходящих в костном мозге, которые мешают производству тромбоцитов, в конечном счете, приводят к нарушению синтеза эритроцитов и лейкоцитов и, следовательно, проявляются в виде разл. аномалий при определении ОАК. Неиммунные нарушения, вызывающие повышенное разрушение тромбоцитов, обычно наблюдаются при серьезных системных заболеваниях с выраженными клиническими проявлениями, такими как ГУС и ДВС-синдром (см. рис. 2 и табл. ниже).
У пациентов, получающих гепарин, может развиться гепарин-индуцированная тромбоцитопения. Изолированное увеличение размеров селезенки указывает на вероятность развития гиперспленизма, вызванного заболеванием печени или тромбозом воротной вены. Аутоиммунная тромбоцитопения м.б. первичным проявлением СКВ, ВИЧ-инфекции, вариабельного неклассифицируемого иммунодефицита и, редко, — лимфомы или аутоиммунного лимфопролиферативного синдрома.
Синдром Вискотта-Олдрича следует рассматривать у молодых мужчин с выявленной тромбоцитопенией и мелкими тромбоцитами, особенно при наличии экземы и рецидивирующей инфекции в анамнезе.
7. Лечение. В настоящее время существует ряд вариантов лечения (табл. 3), однако нет данных, свидетельствующих о том, что лечение может повлиять на краткосрочный или долгосрочный клинический исход ИТП. Несмотря на наличие тяжелой тромбоцитопении, впервые возникшая ИТП проявляется у многих пациентов симптомами легкой степени, только петехиями и пурпурой на коже. По сравнению с контрольной группой, лечение, по-видимому, способно индуцировать более быстрое повышение количества тромбоцитов до теоретически приемлемого уровня >20х109/л, хотя нет данных, указывающих на то, что ранняя терапия предотвращает в/черепное кровоизлияние.
Антитромбоцитарные АТл связываются как с аутологичными тромбоцитами, так и с полученными в результате гемотрансфузии.
Т.о., трансфузия тромбоцитов при ИТП обычно противопоказана, если не наблюдается угрожающего жизни кровотечения. Основные подходы к ведению ИТП включают в себя следующее.
1) Отсутствие терапии, только обучение и консультирования пациента и членов его семьи в случае незначительных, легких и умеренных симптомов, как описано выше. При этом подходе подчеркивается типично доброкачественная природа ИТП, что позволяет избежать резких изменений состояния, которые возникают после начала инвазивной терапии. Такой подход гораздо менее экономически затратен, а побочные эффекты минимальны. Наблюдение рекомендуется Амер. обществом гематологов для детей только с легкими симптомами кровотечения, такими как кровоподтеки или петехии.
2) Терапия в/в-иммуноглобулин или ГКС, особенно для детей с кожно-слизистыми кровотечениями. В методических рекомендациях Амер. общества гематологов указано следующее: «В качестве терапии первой линии следует применять однократную дозу в/в-иммуноглобулина (0,8-1,0 г/кг) или короткий курс ГКС». В/в-иммуноглобулин в дозе 0,8-1,0 г/кг в сутки 1-2 дня индуцирует быстрое повышение количества тромбоцитов (обычно >20х109/л) у 95% пациентов не позднее, чем через 48 ч. В/в-иммуноглобулин, по-видимому, индуцирует ответ, снижая Fc-рецептор-опосредованный фагоцитоз тромбоцитов, покрытых АТл. Терапия в/в-иммуноглобулином является дорогостоящей и занимает много времени.
Кроме того, после инфузии наблюдается высокая частота головных болей и рвоты, что наводит на мысль о в/в иммуноглобулине — индуцированном асептическом менингите.
3) Терапия ГКС уже много лет применяется для лечения острой и хронической форм ИТП у взрослых и детей. Преднизолон в дозе 1-4 мг/кг в сутки, по-видимому, вызывает более быстрое повышение количества тромбоцитов, чем наблюдается у нелеченных пациентов с ИТП. Терапия ГКС обычно назначается короткими курсами до тех пор, пока не будет достигнуто повышение количества тромбоцитов до уровня >20х109/л, чтобы избежать долгосрочных побочных эффектов данной терапии, особенно отставания в росте, СД и остеопороза.
Каждый из этих ЛП м.б. использован для лечения обострений ИТП, которые обычно возникают через несколько нед после первоначального курса терапии. В особом случае развития в/черепного кровоизлияния следует использовать несколько методов лечения, включая гемотрансфузию тромбоцитов, в/в-иммуноглобулин, высокие дозы ГКС, а также оперативно получить консультацию нейрохирурга и общего хирурга.
Относительно методов лечения острой формы ИТП у детей нет единого мнения, за исключением того, что пациенты с массивным кровотечением (<5% детей с ИТП) нуждаются в терапии. В/черепные кровоизлияния встречаются редко, и нет данных, свидетельствующих о том, что терапия на самом деле снижает их частоту. Кровотечение из слизистых оболочек, в частности, вызывает самые большие опасения с точки зрения прогнозирования развития тяжелого кровотечения.
Спленэктомия при ИТП показана в двух случаях: ребенок более старшего возраста (>4 лет) с тяжелой формой ИТП, которая длится >1 года (хроническая ИТП) и симптомами, которые трудно контролировать с помощью терапии, а также при угрожающем жизни кровотечении (в/черепное кровоизлияние), осложняющим острую ИТП, если количество тромбоцитов не м.б. быстро скорректировано с помощью трансфузии тромбоцитов и введения в/в-иммуноглобулин и ГКС. Спленэктомия ассоциируется с пожизненным риском развития подавляющей постспленэктомической инфекции, вызываемой инкапсулированными организмами, повышенным риском развития тромбоза и возможностью развития легочной гипертензии в зрелом возрасте. В качестве альтернативы спленэктомии для лечения хронической формы ИТП у детей применялся ритуксимаб по неутвержденным показаниям.
У 30-40% детей ритуксимаб индуцировал частичную или полную ремиссию. Для повышения количества тромбоцитов также применяются агонисты рецепторов тромбопоэтина, которые одобрены для педиатрического применения.
8. Хроническая аутоиммунная тромбоцитопеническая пурпура. У 20% пациентов с острой формой ИТП наблюдается стойкая тромбоцитопения в течение >12 мес, т.е., как говорится, хроническая ИТП. В этом случае должно быть проведено тщательное повторное обследование для оценки ассоциированных нарушений, особенно по поводу аутоиммунных заболеваний (напр., СКВ), хронических инфекционных заболеваний (напр., ВИЧ), и неиммунных причин хронической тромбоцитопении, таких как тип 2В и тромбоцитарный тип болезни Виллебранда, Х-сцепленная тромбоцитопения, аутоиммунный лимфопролиферативный синдром, синдром вариабельного неклассифицируемого иммунодефицита, аутосомная макротромбоцитопения и синдром Вискотта-Олдрича (также Х-сцепленный). Следует учитывать наличие сопутствующей инфекции Н. pylori и, если она обнаружена, провести ее лечение.
Целью терапии является контроль симптомов и предотвращение серьезных кровотечений. При ИТП селезенка является основным местом как синтеза антитромбоцитарных АТл, так и разрушения тромбоцитов. Спленэктомия вызывает полную ремиссию у 64-88% детей с хронической формой ИТП. Несмотря на благоприятный исход в виде ремиссии, после проведения спленэктомии в течение всей жизни сохраняется риск развития подавляющей постспленэктомической инфекции. На решение о проведении спленэктомии часто влияют вопросы качества жизни пациента, а также насколько просто осуществлять ведение ребенка с помощью ЛС, такой как терапия в/в-иммуноглобулин, ГКС, в/в анти-D иммуноглобулин, или ритуксимабом.
Два эффективных ЛС, стимулирующих тромбоцитопоэз, ромиплостим и элтромбопаг (см. рис. 1) были одобрены FDA для лечения взрослых и детей с хронической формой ИТП.
Хотя эти ЛС не влияют на механизм развития ИТП, повышения количества тромбоцитов м.б. достаточно, чтобы компенсировать их усиленное разрушение, купировать кровотечение и поддерживать количество тромбоцитов на уровне >50x109/л.
г) Тромбоцитопения, индуцированная приемом лекарственных средств. Некоторые ЛП могут спровоцировать иммунную тромбоцитопению в результате либо влияния на иммунные процессы, либо повреждения мегакариоцитов. Распространенные в педиатрии ЛП, вызывающие тромбоцитопению, включают в себя вальпроевую кислоту, фенитоин, карбамазепин, сульфаниламиды, ванкомицин и триметоприм/сульфаметоксазол. Гепарин-индуцированная тромбоцитопения (и иногда ассоциированный тромбоз) редко встречается у педиатрических пациентов, но она развивается в случаях, когда после воздействия гепарина у пациента появляется АТл, направленное против комплекса гепарин-тромбоцитарный фактор 4.
Рекомендуемое лечение гепарин-индуцированной тромбоцитопении включает прямые ингибиторы тромбина, такие как аргатробан или данапароид, и исключение всех источников гепарина, в т.ч. и для промывания магистрали катетера.
д) Разрушение тромбоцитов неиммунного характера. ДВС-синдром, ГУС и тромботическая тромбоцитопеническая пурпура имеют общую гематологическую картину тромботической микроангиопатии, при которой происходит разрушение эритроцитов и наблюдается тромбоцитопения потребления, вызванная отложением тромбоцитов и фибрина в микроциркуляторном русле. Микроангиопатическая гемолитическая анемия характеризуется наличием фрагментов эритроцитов, в т.ч. шлемовидных и шиповидных клеток, шистоцитов и сфероцитов.
е) Гемолитико-уремический синдром. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
ж) Тромботическая тромбоцитопеническая пурпура. Тромботическая тромбоцитопеническая пурпура — редкая форма тромботической микроангиопатии, характеризующаяся пятеркой симптомов: лихорадкой, микроангиопатической гемолитической анемией, тромбоцитопенией, нарушением функции почек и изменениями в ЦНС, клинически сходными с ГУС (табл. 4). Хотя тромботическая тромбоцитопеническая пурпура м.б. врожденной, она обычно встречается у взрослых и иногда у подростков. Микроваскулярные тромбозы ЦНС вызывают малозаметные неврологические отклонения, которые варьируют от изменений эмоциональных реакций и чувства ориентации до афазии, слепоты и судорог.
Начальные проявления часто неспецифичны (слабость, боль, рвота); быстрое распознавание этого расстройства имеет решающее значение. Результаты лабораторных исследований служат важным ориентиром в диагностике и показывают микроангиопатическую гемолитическую анемию, характеризующуюся морфологически аномальными эритроцитами, шизоцитами, сфероцитами, шлемовидными эритроцитами и повышенным количеством ретикулоцитов в сочетании с тромбоцитопенией. Исследования коагуляции обычно не являются диагностическими. Показатели АМК и креатинина в крови иногда повышены. Лечение приобретенной тромботической тромбоцитопенической пурпуры проводится методом плазмафереза (плазмообмена), который эффективен для 80-95% пациентов. Лечение плазмаферезом следует начинать на основании тромбоцитопении и микроангиопатической гемолитической анемии, даже если др. симптомы еще не проявились.
Ритуксимаб, ГКС и спленэктомия назначаются в случае рефракторной формы заболевания. Каплацизумаб, гуманизированный иммуноглобулин против ФВл, блокирует взаимодействие сверхкрупных мультимеров ФВл с тромбоцитами, что во многих случаях приводит к быстрому разрешению острой формы тромботической тромбоцитопенической пурпуры.
Аутоиммунная тромботическая тромбоцитопеническая пурпура м.б. преходящей, рецидивирующей, лекарственно-ассоциированной (тиклопидин, клопидогрел) или наблюдается в некоторых случаях тромботическая тромбоцитопеническая пурпура, ассоциированная с беременностью. Мутации в гене ADAMTS13 часто обусловлены семейными и хроническими формами рецидивирующего респираторного папилломатоза.
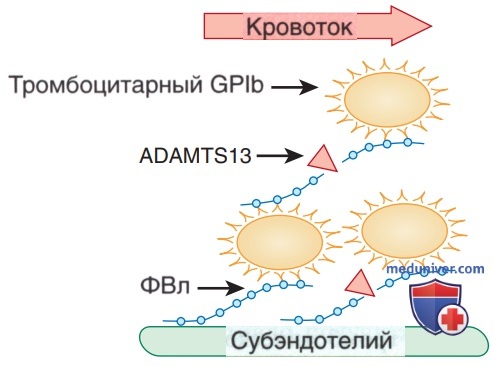
Большинство случаев тромботической тромбоцитопенической пурпуры вызвано аутоАТл-опосредованным дефицитом белка ADAMTS13 (дезинтегрин и металлопротеиназа с мотивом тромбоспондина 1-го типа 13), который отвечает за расщепление высокомолекулярных мультимеров ФВл и, по-видимому, играет ключевую роль в развитии тромботической микроангиопатии (рис. 4). В отличие от тромботической тромбоцитопенической пурпуры, уровни металлопротеиназы при ГУС, как правило, нормальные. Врожденный дефицит металлопротеиназы вызывает редкие семейные случаи тромботической тромбоцитопенической пурпуры / ГУС, обычно проявляющиеся рецидивирующими эпизодами тромбоцитопении, гемолитической анемии и поражения почек, с неврологическими изменениями или без них, которые часто наблюдаются в младенчестве после интеркуррентного заболевания.
Рисунок 4. Патогенез тромботической тромбоцитопенической пурпуры. Мультимеры фактора Виллебранда (ФВл) способствуют адгезии тромбоцитов к субэндотелию, связываясь с экспонированной соединительной тканью, а затем с тромбоцитарным гликопротеином lb (GPIb). В текущей крови напряжение сдвига развертывает сверхкрупные мультимеры ФВл в тромбе, насыщенном тромбоцитами, и позволяет ADAMTS13 расщеплять специфическую связь Tyr-Met во 2-м из 3 доменов А в субъединицах ФВл. Расщепление уменьшает размер мультимеров ФВл и сдерживает рост тромба. В отсутствие ADAMTS13 продолжается зависимая от ФВл концентрация тромбоцитов, что в конечном итоге приводит к микрососудистому тромбозу и тромботической тромбоцитопенической пурпуре
Было установлено, что аномалии системы комплемента также ассоциированы с редкими случаями семейной тромботической тромбоцитопенической пурпуры. Дефицит ADAMTS13 можно лечить с помощью многократных вливаний свежезамороженной плазмы.
з) Синдром Казабаха-Мерритта. Синдром Казабаха-Мерритта — это состояние, характеризующееся наличием гигантской гемангиомы в сочетании с локализованным ДВС, вызывающим тромбоцитопению и гипофибриногенемию. У большинства пациентов локализация гемангиомы не вызывает сомнений, но для выявления забрюшинных и интраабдоминальных гемангиом может потребоваться диагностическая визуализация. Внутри гемангиомы происходит потребление тромбоцитов и активация коагуляции с потреблением фибриногена и образованием продуктов распада фибрина (фибриногена). Артериовенозная мальформация внутри очагов поражения может вызвать СН.
С точки зрения патофизиологии, синдром Казабаха-Мерритта, по-видимому, чаще развивается в результате капошиформной гемангиоэндотелиомы или тафтинговой гемангиомы, а не простой гемангиомы. В мазке периферической крови видны микроангиопатические изменения.
Для лечения синдрома Казабаха-Мерритта применялись разл. методы, включая пропранолол, хирургическое иссечение (при возможности), лазерную фотокоагуляцию, высокие дозы ГКС, местную лучевую терапию, антиангиогенные ЛП, такие как интерферон а-2 и винкристин. Со временем у большинства пациентов, у которых заболевание проявилось в младенческом возрасте, наблюдалась регрессия гемангиомы. В случае ассоциированной коагулопатии эффективным может оказаться пробная антифибринолитическая терапия е-аминокапроновой кислотой или АКТ низкомолекулярным гепарином.
и) Секвестрация. Тромбоцитопения развивается у пациентов с массивной спленомегалией, поскольку селезенка действует как губка для тромбоцитов и секвестрирует их в большом количестве. У большинства таких пациентов в ОАК наблюдается также легкая лейкопения и анемия. Пациенты с тромбоцитопенией, вызванной секвестрацией тромбоцитов в селезенке, должны пройти обследование для диагностики этиологии спленомегалии, которая м.б. обусловлена инфекционными, воспалительными, инфильтративными, неопластическими, обструктивными и гемолитическими нарушениями.
к) Синдромы врожденной тромбоцитопении. См. табл. 2.
Врожденная амегакариоцитарная тромбоцитопения обычно проявляется в течение первых нескольких дней или первой недели жизни, когда у ребенка появляются петехии и пурпура, вызванные глубокой тромбоцитопенией. Врожденная амегакариоцитарная тромбоцитопения вызывается редким дефектом гемопоэза в результате мутации в гене рецептора стволовых клеток (MPL). За исключением патологии кожи и слизистых оболочек, результаты физикального обследования являются нормальными. Исследование костного мозга показывает отсутствие мегакариоцитов. Через какое-то время у таких пациентов часто развивается недостаточность костного мозга (аплазия). Трансплантация гемопоэтических стволовых клеток является радикальным методом терапии.
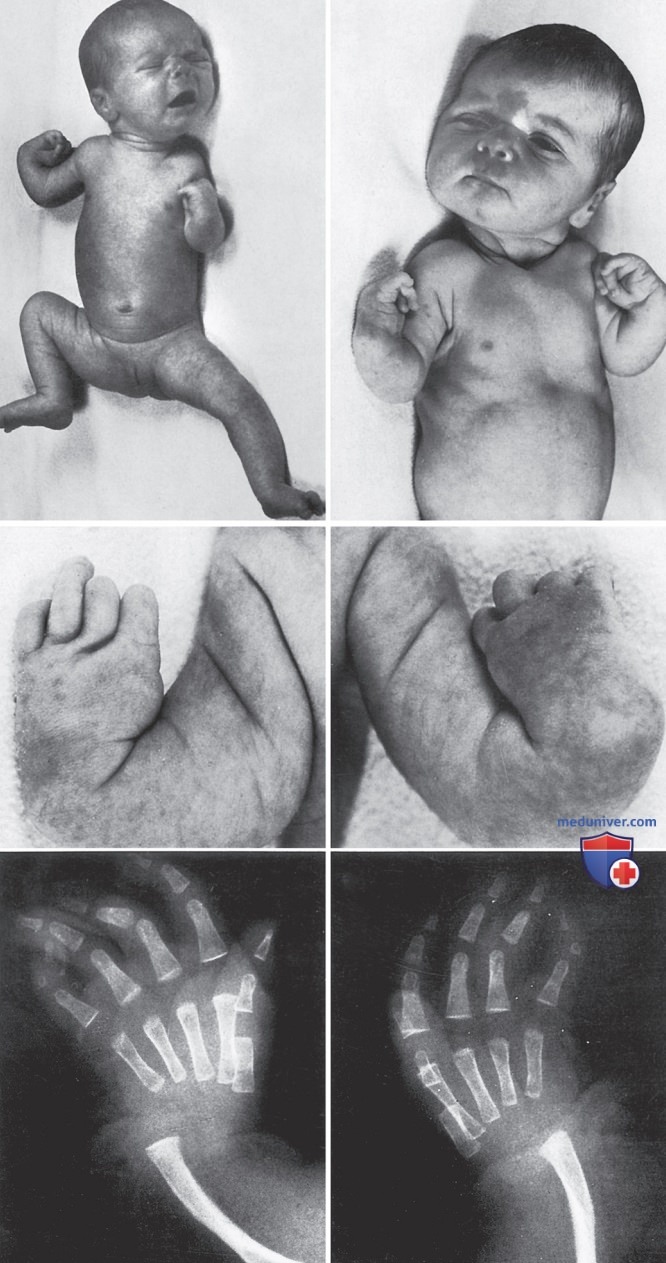
TAR-синдром представляет собой тромбоцитопению (отсутствие или гипоплазию мегакариоцитов), которая проявляется в раннем младенчестве двусторонними аномалиями лучевой кости разл. степени тяжести, варьирующими от легких изменений до выраженного укорочения конечности (рис. 5). У многих таких пациентов также встречаются др. скелетные аномалии локтевой и лучевой костей, а также нижних конечностей. Большие пальцы сохраняются в норме. Непереносимость молочных смесей с коровьим молоком (встречается в 50% случаев) может осложнить лечение, вызвав ЖКК, повышенную тромбоцитопению, эозинофилию и лейкемоидную реакцию. Тромбоцитопения при TAR-синдроме часто переходит в стадию ремиссии в течение первых нескольких лет жизни. Молекулярная основа развития TAR-синдрома связана с мутацией в гене RBM8A.
Рисунок 5. Новорожденный, первый ребенок у молодых, здоровых родителей, с синдромом тромбоцитопении с отсутствием лучевой кости (TAR-синдромом), включающим гиперэозинофилию и анемию. Наблюдаются гипоплазия дистальных отделов плечевого пояса, двусторонняя дисплазия тазобедренного сустава, умеренная деформация пяточной кости и клинодактилия обоих мизинцев. У этого пациента отмечалась выраженная аллергия на коровье молоко с последующей диареей, рвотой, снижением массы тела и количества тромбоцитов, поэтому в обязательном порядке была назначена диета, исключающая употребление коровьего молока. Наблюдается стойкий дефект переносицы и выраженное дугообразное искривление нижних конечностей
У нескольких пациентов наблюдался синдром амегакариоцитарной тромбоцитопении с лучелоктевым синостозом, вызванный мутацией в гене НОХА11. В отличие от TAR-синдрома, эта мутация вызывает аплазию костного мозга.
Синдром Вискотта-Олдрича характеризуется тромбоцитопенией с очень мелкими тромбоцитами, экземой и рецидивирующей инфекцией, обусловленной иммунодефицитом. Синдром Вискотта-Олдрича наследуется как Х-сцепленный рецессивный признак; ген, мутация в котором обусловливает развитие синдрома Вискотта-Олдрича, был секвенирован.
Белок синдрома Вискотта-Олдрича, по-видимому, играет важную роль в регуляции цитоскелетной архитектуры как тромбоцитов, так и Т-лимфоцитов в ответ на рецептор-опосредованную сигнальную систему клетки. Белок синдрома Вискотта-Олдрича экспрессируется во всех клетках гемопоэтической системы. Молекулярное тестирование семей с Х-сцепленной тромбоцитопенией показало, что многие пораженные члены семьи имеют точечную мутацию в гене синдрома Вискотта-Олдрича, тогда как у пациентов с проявлением клинической картины синдрома Вискотта-Олдрича в полном объеме отмечаются крупные делеции генов. Исследование костного мозга при синдроме Вискотта-Олдрича показывает нормальное количество мегакариоцитов, хотя они могут иметь аномальные морфологические характеристики. Продолжительность жизни перелитых тромбоцитов находится в пределах нормы.
Спленэктомия часто корректирует тромбоцитопению, что указывает на ускоренное разрушение тромбоцитов при синдроме Вискотта-Олдрича. Пациенты после спленэктомии подвергаются повышенному риску генерализованной инфекции и на протяжении всей жизни нуждаются в профилактике АБТ против инкапсулированных организмов. У 5-15% пациентов развивается лимфоретикулярная неоплазия. Успешное проведение трансплантации гемопоэтических стволовых клеток приводит к излечению синдрома Вискотта-Олдрича. Х-сцепленная макротромбоцитопения и дизэритропоэз были связаны с мутациями в гене GATA1, эритроидном и мегакариоцитарном факторе транскрипции.
Тромбоцитопения, ассоциированная с мутацией в гене MYH9, объединяет ряд разнообразных наследственных тромбоцитопенических синдромов (напр., синдромы Себастьяна (Sebastian), Эпштейна (Epstein), Мэй-Хегглина (May, Richard; Hegglin, Robert), Фехтнера (Fechtner)), характеризующихся АуД макротромбоцитопенией, включениями нейтрофилов и разл. физическими аномалиями, в т.ч. нейросенсорной потерей слуха и заболеваниями почек и глаз. Эти нарушения были вызваны разл. мутациями в гене MYH9 (немышечная тяжелая цепь миозина-IIа). Тромбоцитопения обычно протекает в легкой форме и не прогрессирует. У некоторых др. пациентов с рецессивно унаследованной макротромбоцитопенией отмечаются аномалии в хромосоме 22q11. Мутации в гене гликопротеина Ibβ, важнейшего компонента тромбоцитарного рецептора ФВл, могут привести к развитию синдрома Бернара-Сулье (Bernard Jean; Soulier Jean Pierre).
л) Неонатальная тромбоцитопения. Тромбоцитопения у новорожденных редко свидетельствует о первичном нарушении мегакариопоэза. Обычно такая тромбоцитопения развивается в результате системного заболевания или переноса материнских АТл, направленных против тромбоцитов плода (см. табл. 2). Неонатальная тромбоцитопения часто возникает в связи с врожденными вирусными инфекциями, главным образом краснухой, ЦМВ, протозойными инфекциями (напр., токсоплазмозом) и сифилисом, а также перинатальными бактериальными инфекциями, особенно вызванными гр/о бактериями. Тромбоцитопения, связанная с ДВС-синдромом, м.б. причиной тяжелого спонтанного кровотечения. Констелляция выраженной тромбоцитопении и аномалий БП часто встречается при некротизирующем энтероколите и др. нарушениях, вызывающих некроз кишечника.
При развитии тромбоцитопении у больного ребенка необходимо как можно быстрее провести исследование на выявление вирусных или бактериальных патогенных микроорганизмов.
АТл-опосредованная тромбоцитопения у новорожденного возникает из-за трансплацентарного переноса к плоду материнских АТл, направленных против тромбоцитов плода. Аллоиммунная тромбоцитопеническая пурпура новорожденных вызывается выработкой материнских АТл против АГн, присутствующих на тромбоцитах плода и отца, которые распознаются материнской иммунной системой как чужеродные. Это тромбоцитарный эквивалент Rh-конфликта у новорожденного. Заболеваемость аллоиммунной тромбоцитопенической пурпурой новорожденных составляет 1:4000-5000 живорождений. Клинические проявления аллоиммунной тромбоцитопенической пурпуры новорожденных такие же, как у внешне здорового ребенка, у которого в течение первых нескольких дней после родов развиваются генерализованные петехии и пурпура.
Лабораторные исследования показывают нормальное количество тромбоцитов у матери, но умеренную или тяжелую тромбоцитопению у новорожденного.
В тщательно собранном анамнезе не должно быть данных о тромбоцитопении у матери. Вплоть до 30% младенцев с тяжелой формой аллоиммунной тромбоцитопенической пурпуры новорожденных могут развивать в/черепное кровоизлияние как в пренатальном, так и в перинатальном периоде. В отличие от Rh-конфликта, при первой беременности может развиться тяжелая форма аллоиммунной тромбоцитопенической пурпуры новорожденных. При последующих беременностях аллоиммунная тромбоцитопеническая пурпура новорожденных м.б. еще более тяжелой, чем при первой беременности.
Диагноз аллоиммунной тромбоцитопенической пурпуры новорожденных ставится на основании тестирования на наличие материнских аллоАТл, направленных против АГн тромбоцитов отца. Для определения целевого аллоАГн м.б. проведены специальные исследования. Наиболее распространенной причиной является несовместимость родителей по аллоАГн тромбоцитов НРА-1а. Были определены специфические полиморфизмы последовательностей ДНК, которые позволяют проводить информативное пренатальное тестирование для выявления беременных женщин из группы риска. ДД аллоиммунной тромбоцитопенической пурпуры новорожденных направлена на исключение трансплацентарного переноса материнских антитромбоцитарных аутоАТл к плоду (материнской ИТП) и более распространенной вирусной или бактериальной инфекции.
Лечение аллоиммунной тромбоцитопенической пурпуры новорожденных требует введения в/в-иммуноглобулин матери в пренатальный период. Терапия обычно начинается во втором триместре и продолжается на протяжении всей беременности. Количество тромбоцитов плода можно контролировать посредством чрескожного забора пуповинной крови. Роды должны проводиться путем кесарева сечения. После родов, при сохранении тяжелой тромбоцитопении, трансфузия 1 единицы тромбоцитов, имеющих материнские аллоАГн (напр., отмытые материнские тромбоциты), вызовет повышение уровня тромбоцитов, что будет способствовать успешному гемостазу. Тем не менее гораздо проще, скорее всего, будет провести гемотрансфузию тромбоцитов от случайного донора. В некоторых медцентрах могут отсутствовать наиболее часто встречающиеся АГн.
После рождения одного больного ребенка очень важно провести генетическое консультирование, чтобы проинформировать родителей о высоком риске тромбоцитопении при последующих беременностях.
Дети, рожденные от матерей с ИТП, по-видимому, имеют более низкий риск серьезных кровотечений, чем дети, рожденные с аллоиммунной тромбоцитопенической пурпурой новорожденных, хотя у первых может возникнуть тяжелая тромбоцитопения. Количество тромбоцитов у матери до беременности может иметь некоторую прогностическую ценность т.к. тяжелая форма материнской тромбоцитопении до родов, по-видимому, связана с более высоким риском развития тромбоцитопении у плода. У матерей, перенесших спленэктомию по поводу ИТП, уровень тромбоцитов м.б. нормальным и, т.о., не иметь прогностического значения относительно тромбоцитопении у плода.
Лечение включает введение ГКС матери в пренатальный период и в/в-иммуноглобулин, а иногда и ГКС ребенку после родов. Тромбоцитопения у младенца, развившаяся и в результате аллоиммунной тромбоцитопенической пурпуры новорожденных, и в результате материнской ИТП, обычно проходит в течение 2-4 мес после родов. Наибольший риск возникает непосредственно в перинатальный период.
В период новорожденности часто встречаются два синдрома врожденной недостаточности продукции тромбоцитов. При врожденной недостаточности продукции тромбоцитов у новорожденного вскоре после рождения проявляются петехии и пурпура. Результаты физикального обследования в остальном соответствуют норме. В костном мозге отсутствуют мегакариоциты. Этот синдром вызывается мутацией в гене мегакариоцитарного рецептора, который необходим для развития всех линий гемопоэтических клеток. Со временем развивается панцитопения, а радикальным лечением является трансплантация гемопоэтических стволовых клеток. TAR-синдром представляет собой тромбоцитопению, которая проявляется в раннем младенчестве, с двусторонними аномалиями лучевой кости разл. степени тяжести, варьирующимися от легких изменений до выраженного укорочения конечностей. Этот синдром часто переходит в стадию ремиссии спустя несколько первых лет жизни (рис. 4).
м) Тромбоцитопения вследствие приобретенных нарушений, вызывающих снижение продукции тромбоцитов. Заболевания костного мозга, при которых происходит ингибирование мегакариоцитопоэза, обычно влияют и на производство эритроцитов и лейкоцитов. При инфильтративных заболеваниях, включающих ЗНО, такие как острый лимфоцитарный лейкоз, гистиоцитоз, лимфомы и болезнь депонирования, обычно наблюдаются либо отклонения при физикальном обследовании (лимфаденопатия, гепатоспленомегалия или ЗНО), либо отклонения в количестве лейкоцитов, либо анемия. Апластические процессы могут проявляться как изолированная тромбоцитопения, хотя обычно ОАК показывает и др. признаки (лейкопению, нейтропению, анемию или макроцитоз).
У детей с конституциональной апластической анемией (анемией Фанкони (Fanconi, Guido)) часто, но не всегда, при обследовании наблюдаются разл. аномалии, включая аномалии лучевой кости, др. аномалии скелета, низкий рост, микроцефалию и гиперпигментацию. Исследование костного мозга следует проводить, если тромбоцитопения ассоциируется с аномалиями, обнаруженными при физикальном обследовании или при исследовании др. линий клеток крови.
н) Нарушения функции тромбоцитов. Пока не существует простого и достоверного теста для выявления нарушений функции тромбоцитов. В прошлом применялись тесты на время кровотечения и анализ функции тромбоцитов, но ни один из них не обладает достаточной чувствительностью и специфичностью, чтобы подтвердить или исключить дефект тромбоцитов. Тест на время кровотечения измеряет взаимодействие тромбоцитов со стенкой кровеносного сосуда и, т.о., результаты этого теста зависят и от количества тромбоцитов, и от их функции. Прогностическая ценность теста на время кровотечения вызывает сомнения, поскольку время кровотечения зависит от ряда др. факторов, включая мастерство лаборанта и степень сотрудничества пациента, что часто проблематично в случае младенцев или маленьких детей.
Анализ функции тромбоцитов измеряет адгезию и агрегацию тромбоцитов в цельной крови при высокой скорости сдвига, когда кровь подвергается воздействию либо коллагена и адреналина, либо коллагена и АДФ. Результаты оцениваются как время закрытия в секундах. Применение анализа функции тромбоцитов в качестве скринингового остается спорным, как и тест на время кровотечения, из-за низкой специфичности. Пациентам с положительным анамнезом кровотечения, указывающего на болезнь Виллебранда или дисфункцию тромбоцитов, следует проводить специфическое тестирование на ФВл и исследование функции тромбоцитов, независимо от результатов теста на время кровотечения или анализ функции тромбоцитов.
В клинических лабораториях функция тромбоцитов измеряется методом агрегометрии тромбоцитов. Агонисты, такие как коллаген, АДФ, ристоцетин, адреналин, арахидоновая кислота и тромбин (или пептид рецептора тромбина), добавляются в агрегометре в насыщенную тромбоцитами плазму, и через некоторое время агрегация тромбоцитов измеряется автоматически. В то же время др. приборы измеряют высвобождение гранулированного содержимого, такого как АТФ, из тромбоцитов после их активации. Способность тромбоцитов к агрегации и их метаболическая активность м.б. оценены одновременно. При исследовании пациента на предмет возможной дисфункции тромбоцитов крайне важно исключить наличие др. экзогенных агентов, а также исключить по возможности прием всех ЛП в течение 2 нед. Для постановки более точного диагноза часто необходима дальнейшая оценка методом проточной цитометрии или молекулярным методом.
о) Приобретенные нарушения функции тромбоцитов. Ряд системных заболеваний ассоциируется с дисфункцией тромбоцитов, чаще всего это заболевания печени, почек (уремия) и нарушения, которые вызывают повышение количества продуктов деградации фибрина. Такие нарушения часто вызывают длительное кровотечение и нередко связаны с др. нарушениями механизма свертывания крови. Наиболее важным аспектом ведения такого нарушения является лечение первичного заболевания. В случаях, когда лечение первичного процесса не представляется возможным, инфузии десмопрессина успешно усиливают гемостаз и корректируют показатели времени кровотечения. У некоторых пациентов трансфузии тромбоцитов и криопреципитата также эффективно улучшали гемостаз.
Многие ЛП изменяют функцию тромбоцитов. Ацетилсалициловая кислота является наиболее распространенным среди взрослых пациентов ЛП, изменяющим функцию тромбоцитов. Ацетилсалициловая кислота («Аспирин») необратимо ацетилирует фермент ЦОГ, которая играет решающую роль в образовании тромбоксана А2. Ацетилсалициловая кислота обычно вызывает умеренную дисфункцию тромбоцитов, которая становится более выраженной при наличии др. аномалии системы гемостаза. ЛП, которые влияют на функцию тромбоцитов и часто назначаются детям, включают в себя др. НПВП, вальпроевую кислоту и высокие дозы пенициллина. Специфические ЛС для терапевтического ингибирования функции тромбоцитов — это ЛП, которые блокируют тромбоцитарный рецептор АДФ (клопидогрел) и антагонисты рецептора aIIb-β3, а также ацетилсалициловая кислота.
п) Врожденные нарушения функции тромбоцитов. Тяжелые нарушения функции тромбоцитов обычно проявляются петехиями- и пурпурой вскоре после рождения, особенно после вагинальных родов. Дефекты тромбоцитарного комплекса GPIb (рецептор ФВл) или комплекса aIIb-β3 (рецептор фибриногена) вызывают врожденное нарушение функции тромбоцитов тяжелой степени. Хотя существуют лабораторные тесты на определение функции тромбоцитов, в настоящее время быстро развивается метод молекулярно-генетического тестирования для выявления заболеваний тромбоцитов.
Синдром Бернара-Сулье (Bernard, Jean; Soulier, Jean Pierre), тяжелое врожденное нарушение функции тромбоцитов, вызывается отсутствием или тяжелой формой дефицита рецепторов ФВл на мембране тромбоцитов. Этот синдром характеризуется тромбоцитопенией с гигантскими тромбоцитами и значительно пролонгированным временем кровотечения (>20 мин) или временем закрытия анализа функции тромбоцитов.
У пациентов могут наблюдаться множественные кровоизлияния в кожу, слизистые и ЖКК. Тесты на агрегацию тромбоцитов показывают отсутствие ристоцетин-индуцированной агрегации тромбоцитов, но агрегация со всеми др. агонистами определяется в норме. Ристоцетин индуцирует связывание ФВл с тромбоцитами и агглютинирует тромбоциты. Тестирование на ФВл дает нормальные результаты. Комплекс GPIb взаимодействует с цитоскелетом тромбоцитов; считается, что нарушение этого взаимодействия и является причиной большого размера тромбоцитов. Синдром Бернара-Сулье наследуется как АуР-заболевание. Генетические мутации, вызывающие этот синдром, как правило, затрагивают гены, отвечающие за образование комплекса GPIb из гликопротеинов Ibα, Ibβ, V и IX.
Тромбастения Гланцмана (Glanzmann, Eduard) — это врожденное заболевание, ассоциированное с нарушением функции тромбоцитов тяжелой степени, при котором наблюдается увеличение время кровотечения и нормальное количество тромбоцитов. Размер и морфологические характеристики тромбоцитов в мазке периферической крови находятся в норме, а время закрытия анализа тромбоцитов или время кровотечения значимо нарушены. Исследования агрегации показывают нарушения или отсутствие агрегации со всеми используемыми агонистами, кроме ристоцетина, поскольку ристоцетин агглютинирует тромбоциты и не требует метаболически активных тромбоцитов. Это нарушение вызывается дефицитом тромбоцитарного рецептора фибриногена aIIb-β3, основного интегринового комплекса на поверхности тромбоцитов, который меняет конформацию посредством сигнального пути «изнутри-наружу» при активации тромбоцитов.
Фибриноген связывается с этим комплексом при активации тромбоцитов и вызывает их агрегацию. Тромбастения Гланцмана обусловлена идентифицируемыми мутациями в генах aIIb или β3 и наследуется АуР-способом. Как при синдроме Бернара-Сулье, так и при тромбастении Гланцмана диагноз подтверждается методом проточной цитометрии тромбоцитарных гликопротеинов пациента. Кровотечения при тромбастении Гланцмана м.б. довольно тяжелыми и, как правило, в кожу, из слизистых оболочек, включая носовые, гингивальные и ЖКК. Имеются сообщения о радикальном лечении с помощью трансплантации стволовых клеток.
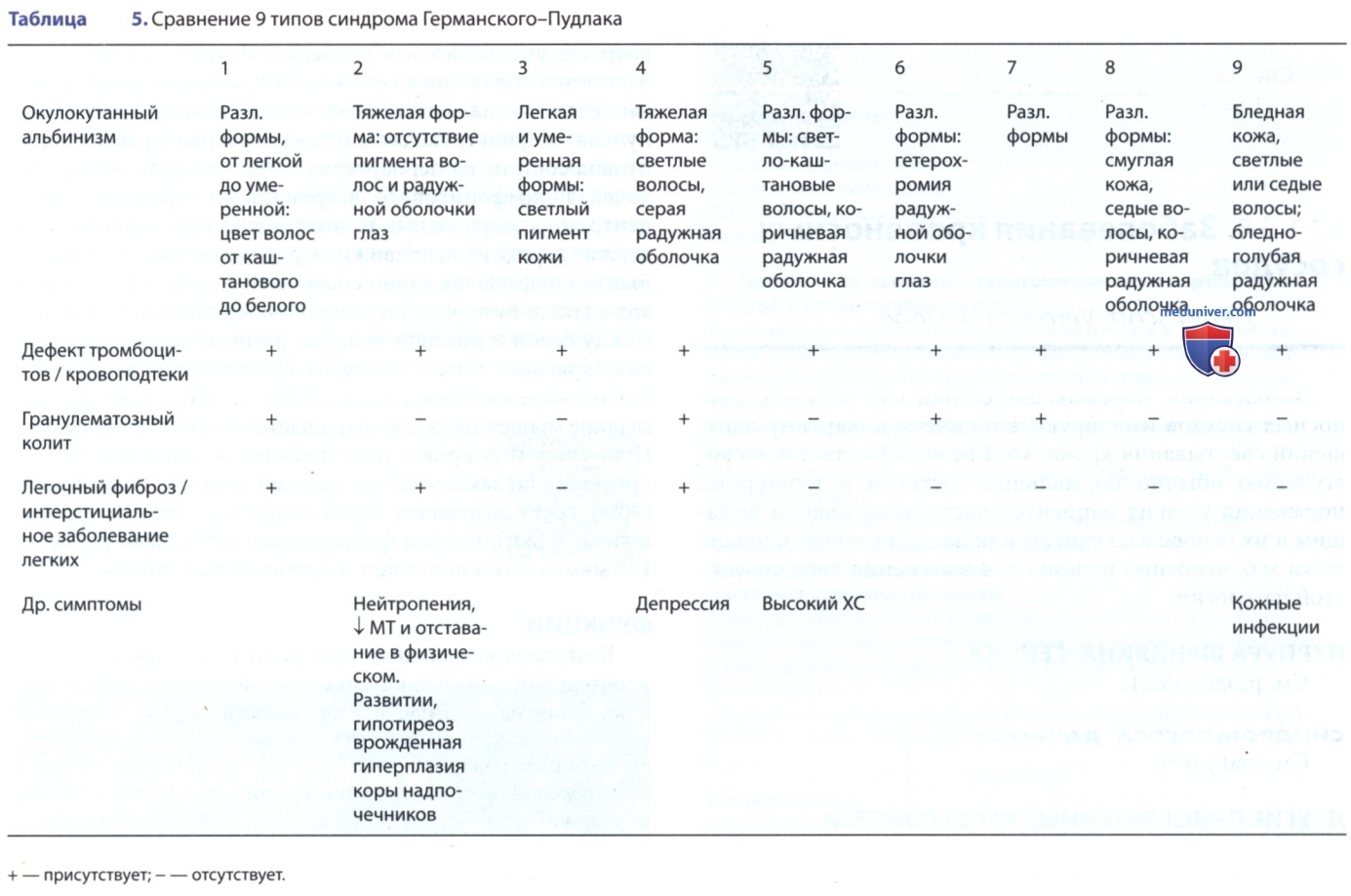
Наследственный дефицит гранул хранения (накопительных вакуолей) тромбоцитов наблюдается при двух хорошо описанных, но редко встречающихся синдромах, которые включают дефицит интрацитоплазматических гранул. Дефицит плотных гранул характеризуется отсутствием гранул, содержащих АДФ, АТФ, Са2+ и серотонин. Это расстройство диагностируется на основании результатов исследования агрегации тромбоцитов, при которых не происходит высвобождение АТФ, и в идеале на основании электронно-микроскопических исследований. Синдром Германского-Пудлака (включая 9 подтипов) — это дефицит плотных гранул, вызванный нарушениями лизосомального хранения. У пораженных пациентов наблюдается окулокутанный альбинизм и кровотечения, вызванные дефектом тромбоцитов; у некоторых пациентов также развивается гранулематозный колит, напоминающий болезнь Крона или легочный фиброз / интерстициальное заболевание легких (табл. 5).
Синдром Чедиака-Хигаси (Chediak Ahuayda, Moises; Higashi, Ototaka) также характеризуется дефектом плотных гранул, иммунной дисфункцией и альбинизмом. Синдром серых тромбоцитов вызывается отсутствием а-гранул тромбоцитов, в результате чего образуются крупные тромбоциты, которые кажутся серыми при окрашивании периферической крови красителем Райта. При этом редком синдроме агрегация и высвобождение тромбоцитов отсутствуют с большинством агонистов, кроме тромбина и ристоцетина. Диагноз подтверждается на основании электронно-микроскопических исследований. АуР-синдром серых тромбоцитов ассоциирован с дефектами гена NBEAL2, в то время как АуД-заболевание ассоциировано с мутацией в гене GFI1B. Тромбоцитарный синдром Квебекский (Quebec) обусловлен деградацией а-гранул тромбоцитов, вызванной дефектами в гене активатора плазминогена урокиназного типа. Лечение обычно включает антифибринолитическую терапию.
1. Другие наследственные нарушения функции тромбоцитов. Аномалии сигнальных путей / активации тромбоцитов и высвобождения гранулярного содержимого вызывают гетерогенную группу нарушений функции тромбоцитов, которые обычно проявляются в виде повышенной склонности к образованию кровоподтеков, носовых кровотечений и меноррагии. Симптомы м.б. едва заметными и часто становятся более выраженными во время операций высокого риска, таких как тонзиллэктомия или аденоидэктомия, или при приеме НПВП. При лабораторном исследовании время кровотечения варьирует, а время закрытия, измеряемое при помощи анализа тромбоцитов, часто, но не всегда, удлинено. Исследования агрегации тромбоцитов показывают недостаточную агрегацию с 1 или 2 агонистами и/или аномальное высвобождение гранулярного содержимого.
Образование тромбоксана из арахидоновой кислоты после активации фосфолипазы имеет решающее значение для нормальной функции тромбоцитов. Дефицит или дисфункция ферментов, таких как ЦОГ и тромбоксансинтаза, которые метаболизируют арахидоновую кислоту, вызывает нарушение функции тромбоцитов. Тромбоциты у таких пациентов не агрегируют в ответ на воздействие арахидоновой кислоты.
Наиболее распространенными нарушениями функции тромбоцитов являются нарушения, которые характеризуются варьирующимися показателями времени кровотечения / времени закрытия анализа тромбоцитов и аномальной агрегацией с 1 или 2 агонистами, обычно АДФ и/или коллагеном. У некоторых из таких пациентов отмечается только сниженное высвобождение АТФ из цитоплазматических гранул; значение этого наблюдения все еще обсуждается.
2. Лечение больных с нарушением функции тромбоцитов. Успешное лечение зависит от степени тяжести как диагноза, так и геморрагического события. При всех нарушениях, кроме нарушений функции тромбоцитов тяжелой степени, во время эпизодов кровотечения легкой и умеренной степени может применяться десмопрессин в/в 0,3 мкг/кг. Помимо того, что десмопрессин повышает уровни ФВл и фактора VIII, он также корректирует время кровотечения и усиливает гемостаз у многих пациентов с легкими и умеренными нарушениями функции тромбоцитов. При кровотечениях из слизистых м.б. эффективна антифибринолитическая терапия. Для пациентов с синдромом Бернара-Сулье или тромбастенией Гланцмана трансфузия тромбоцитов в дозе 0,5-1 ед. тромбоцитов от одного донора корректирует нарушение гемостаза и может даже спасти жизнь.
В редких случаях АТл вырабатываются к дефицитному белку тромбоцитов, что делает пациента невосприимчивым к переливаемым тромбоцитам.
Для таких пациентов эффективным было применение рекомбинантного фактора VIIa, этот метод лечения проходит в настоящее время клинические испытания. При обоих заболеваниях радикальной терапией является трансплантация гемопоэтических стволовых клеток.
р) Заболевания кровеносных сосудов. Заболевания, поражающие стенки или каркас кровеносных сосудов имитируют клиническую картину нарушений свертывания крови, хотя результаты тестов на коагуляцию обычно нормальные. Петехии и пурпурные поражения у таких пациентов часто объясняются лежащим в их основе васкулитом или васкулопатией. Биопсия кожи м.б. особенно полезна для выяснения типа сосудистой патологии.
1) Пурпура Шенляйна-Геноха. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
2) Синдром Элерса-Данлоса. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
3) Другие приобретенные расстройства. Цинга, длительная терапия ГКС и тяжелая недостаточность питания ассоциируются с ослаблением коллагенового матрикса, поддерживающего кровеносные сосуды. Поэтому эти факторы связаны с легким образованием кровоподтеков, и, особенно в случае цинги, кровоточивостью десен и при расшатывании зубов. Поражения кожи, которые первоначально кажутся петехиями и пурпурой, могут наблюдаться при синдромах васкулита, таких как СКВ.