а) Мальформации лимфатической системы. Мальформации лимфатической системы (МЛС) м.б. локализованными или генерализованными, могут ассоциироваться с рядом синдромов, а также развиваться как мальформации, комбинированные с др. типами поражения сосудов [см. классификацию Международного общества по изучению сосудистых аномалий (ISSVA; англ. International Society for the Study of Vascular Anomalies)].
МЛС представляют собой расширенные лимфатические каналы или кисты, выстланные лимфатическими эндотелиальными клетками. МЛС обычно классифицируются в зависимости от размера кисты (макрокистозные, микрокистозные, смешанные) и чаще встречаются в области головы, шеи и подмышечной впадины.
Генерализованная лимфатическая аномалия представляет собой мультифокальную МЛС, которая поражает мягкие ткани, внутренние органы БП и ГК, и часто даже кости. Поражение костей при генерализованной лимфатической аномалии обычно не является прогрессирующим и, как правило, не затрагивает кортикальный слой. Могут возникать хилезные выпоты в плевральные, перикардиальные и перитонеальные области.
Болезнь Горхема-Стаута (болезнь «исчезающей кости») характеризуется МЛС, включающей поражение одной или нескольких костей и приводящей к прогрессирующему разрушению кортикальной костной ткани. МЛС часто поражают мягкие ткани, прилегающие к кости, что также приводит к выпотам при болезни Горхема-Стаута.
Аномалии центральных проводящих лимфатических сосудов в зависимости от пораженной области, могут приводить к хилотораксу, легочной лимфангиэктазии, хилезному асциту, энтеропатии с белковой дистрофией, кожным изменениям, хилезной утечке и изменениям в костях из-за расширения в/костных лимфатических каналов.
Кроме того, МЛС ассоциируются с соматическими мутациями в гене PIK3CA, которые часто также приводят к др. сосудистым мальформациям и регионарной гипертрофии ткани.
1. Генетика. Соматические, гиперактивирующие мутации в гене PIK3CA встречаются при большинстве локализованных МЛС и при МЛС, являющихся частью синдрома, с низкой частотой (<10%). Эти мутации представляют собой те же самые мутации гена PIK3CA, которые встречаются при развитии многих раковых опухолей у человека.
До сих пор неясно, как одни и те же соматические мутации вызывают такое разнообразие фенотипов. Однако активация гена PIK3CA приводит к усилению передачи сигналов по пути AKT/mTOR, что, вероятно, объясняет чувствительность большинства МЛС к сиролимусу.
2. Лечение. Выбор метода лечения МЛС зависит от анат. расположения мальформации, степени поражения органов, а также от выраженности симптомов заболевания. Рекомендуется направление в специализированную клинику сосудистых аномалий, врачи которой обладают опытом и знаниями для принятия соответствующих решений по выбору методов визуализации и лечения.
При локализованных макрокистозных МЛС интервенционная радиология с введением склерозирующих агентов (ОК432, этанол, блеомицин) является наиболее эффективным. Для аномалий, поражающих кожу и слизистые оболочки, может применяться лазерная терапия.
Была доказана эффективность сиролимуса, ингибитора мишени рапамицина в клетках млекопитающих (mTOR), при применении отдельно или в комбинации с интервенционной радиологией при сложных или обширных формах МЛС. Пропранолол был эффективен в некоторых случаях.
б) Лимфангиэктазия. Лимфангиоэктазия больше не рассматривается как отдельное поражение, а входит в группу либо генерализованной лимфатической аномалии, либо МЛС канального (руслового) типа в соответствии с классификацией ISSVA.
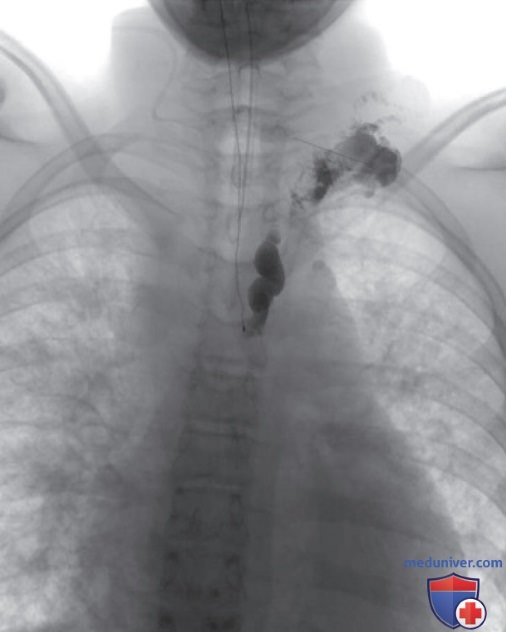
МЛС канального типа может возникать в результате гипоплазии цистерн грудного протока, ЛУ или центральных собирающих протоков, приводя к обструкции лимфотока от поверхностных к центральным собирающим каналам, а также к расширению поверхностных сосудов (рис. ниже). Симптомы зависят от степени обструкции и могут включать в себя энтеропатию с белковой дистрофией при поражении брыжейки или хилезные выпоты, если обструкция происходит в выше.
Интранодальная лимфангиограмма у 15-летнего мальчика с аномалией центральных проводящих лимфатических сосудов и рецидивирующими перикардиальными выпотами. В проходимой верхней части грудного протока наблюдаются застойные явления. Прямая пункция терминального отдела демонстрирует выраженное расширение без самопроизвольного опорожнения
в) Лимфедема. Лимфедема — это локализованный отек, вызванный нарушением лимфотока, который м.б. первичным (врожденным) или приобретенным. Первичные лимфедемы входят в группу МЛС, поскольку они являются результатом дисгенеза лимфатических сетей в период раннего развития.
Первичная лимфедема м.б. обнаружена при синдроме Тернера, синдроме Нунан и болезни Милроя, наследуемой по АуД-типу, и при др. хромосомных аномалиях. Мутации в нескольких генах, включая ген рецептора сосудистого эндотелиального фактора роста-3 (VEGFR3), гены GJC2, PTPN14 и GATA2 ассоциированы с первичной лимфедемой.
Болезнь Милроя вызывается АуР и доминантными мутациями или мутации de novo в гене VEGFR3. Мутации в др. генах связаны со специфическими синдромами: в гене ССВЕ1 — с синдромом Хеннекама, в гене FOXC2 — с синдромом лимфедемы-дистихиаза, в гене SOX18 — с синдромом гипотрихоза-лимфедемы-телеангиэктазии, в генах KMT2D/ MLL2 и KDM6A — с синдромом Кабуки. Односторонняя или двусторонняя лимфедема нижних конечностей у подростка может оказаться болезнью Мейжа.
Приобретенная обструкция лимфотока м.б. обусловлена опухолью, пострадиационным фиброзом и поствоспалительным рубцеванием. В Африке, Азии и Латинской Америке лимфедема нередко обусловлена филяриозом. Одна треть из 120 млн инфицированных людей (в основном это подростки и взрослые) страдает лимфедемой или водянкой оболочек яичка. Повреждение крупных лимфатических сосудов может вызвать скопление лимфатической жидкости в БП (хилезный асцит) или ГК (хилоторакс).
Нелеченная лимфедема м.б. инвалидизирующей и ассоциируется с дисфункцией иммунной системы, воспалением, фиброзом, гипертрофией ПЖК и лимфангиосаркомой. Современные методы лечения направлены на уменьшение местных отеков с помощью массажа, физических упражнений и компрессии. Селен оказался эффективным вспомогательным средством в дополнение к физиотерапии у некоторых взрослых пациентов после лечения рака МЖ.
г) Лимфангиолейомиоматоз. Лимфангиолейомиоматоз характеризуется пролиферацией лимфатических эндотелиальных клеток и гладкомышечных клеток в легких, что приводит к ОДП и лимфотока, образованию кист, пневмотораксу и ДН. На ранних стадиях заболевания его можно принять за БА.
Лимфангиолейомиоматоз встречается у молодых женщин и в трети случаев ассоциируется с мутациями в гене опухолевого супрессора туберозного склероза TSC2. Сиролимус стабилизирует функцию легких, уменьшает симптомы и улучшает качество жизни. Может потребоваться трансплантация легких.
д) Лимфангит. Лимфангит — это воспаление лимфатических сосудов, которые отводят лимфу от инфицированной области. Лимфангит характеризуется болезненными эритематозными полосами на коже, которые отходят проксимально от инфицированного участка. Регионарные ЛУ также м.б. болезненными. Стрептококки группы А и Staphylococcus aureus являются наиболее распространенными патогенами, а терапия должна включать АБ, которые действуют против этих микроорганизмов.