Панцитопения может являться результатом уменьшения ниже нормальных значений показателей всех 3 линий периферической крови: лейкоцитов, тромбоцитов и эритроцитов. Выявление этиологии панцитопении обычно требует микроскопического исследования мазка периферической крови, а также биопсии и аспирата костного мозга для оценки общей морфологии и клеточности. Три основные категории панцитопении связаны с патологиями костного мозга и часто м.б. дифференцированы на основе результатов исследования костного мозга.
Панцитопения с гипоцеллюлярным костным мозгом при биопсии наблюдается при синдромах врожденной недостаточности костного мозга с панцитопенией, при приобретенных апластических анемиях разл. этиологии, а также при гипопластическом варианте миелодиспластического синдрома. Панцитопения с целлюлярным костным мозгом наблюдается при первичных заболеваниях костного мозга (напр., острый лейкоз, миелодисплазия) и при вторичных аутоиммунных расстройствах (напр., аутоиммунный лимфопролиферативный синдром, СКВ), а также при дефиците витамина В12 или фолиевой кислоты, при болезнях депонирования (напр., болезнь Гоше, Ниманна-Пика), при генерализированных инфекциях, саркоидозе и гиперспленизме.
Панцитопения с инфильтрацией костного мозга может наблюдаться при метастатических солидных опухолях, миелофиброзе, гемофагоцитарном лимфогистиоцитозе и остеопорозе. Важно отметить, что существуют исключения из данной классификации. Напр., на ранних стадиях проявления заболевания или в случаях развития миелодиспластического синдрома при синдроме врожденной недостаточности костного мозга может наблюдаться нормоклеточный или гиперклеточный костный мозг.
Врожденные панцитопении с гипоклеточным костным мозгом — это синдром врожденной недостаточности костного мозга, которые характеризуются снижением продукции костным мозгом трех основных гемопоэтических линий, что передается по наследству и приводит к анемии, нейтропении и тромбоцитопении. Следует отметить, что при проявлении симптомов заболевания у пациентов может наблюдаться однолинейная или билинейная цитопения, которая с течением времени может постепенно прогрессировать в панцитопению.
Все расстройства, для которых была расшифрована генетическая основа, до сих пор считаются моногенными. Мутантные гены, согласно законам Менделя, могут передаваться АуД или АуР-путем, или быть сцепленными с Х-хромосомой (табл. 1). Гены-модификаторы и приобретенные факторы также могут оказывать воздействие. Врожденные панцитопении составляют 30% случаев недостаточности костного мозга у детей. Анемия Фанкони считается наиболее распространенной из врожденных панцитопений.
а) Анемия Фанкони:
1. Этиология и эпидемиология. Анемия Фанкони — редкое мультисистемное наследственное заболевание, приводящее к развитию недостаточности костного мозга у больных и предрасположенности к ЗНО, включая миелодисплазию, острый миелоидный лейкоз и карциномы. Пациенты с анемией Фанкони часто имеют врожденные пороки развития и высокую чувствительность к алкилирующим агентам и радиации. Предполагаемая частота анемии Фанкони — 1:200 000 населения в большинстве популяций, но выше среди ашкеназских евреев (1:30 000) и африканеров (1:22 000).
Частота гетерозиготности по анемии Фанкони составляет 1:200-300 в большинстве популяций. На сегодняшний день имеются сообщения, что мутации 21 генов, которые обозначаются как гены FANC, вызывают анемии Фанкони или заболевания, подобные анемии Фанкони. Все эти мутации, за исключением одной, наследуются АуР-путем. Одна необычная форма — рецессивна и сцеплена с Х-хромосомой. Анемия Фанкони встречается во всех расовых и этнических группах. При проявлении заболевания у пациентов могут наблюдаться типичные соматические патологии и аномальные гематологические показатели (у большинства пациентов), физические характеристики м.б. в норме, но гематологические показатели м.б. аномальными (ок. 1/3 пациентов) или могут наблюдаться соматические патологии при нормальных гематологических показателях (процент пациентов не установлен).
Клиническая и гематологическая картина м.б. совершенно разной у пораженных братьев и сестер, даже у монозиготных близнецов.
2. Патология. Все гены анемии Фанкони кодируют белки, которые принимают участие в разл. клеточных путях и играют наиболее заметную роль в поперечной сшивке и репарации ДНК. У пациентов с анемией Фанкони наблюдается дефектная репарация ДНК и повышенная ломкость хромосом, вызванная межнитевыми сшивающими агентами ДНК, такими как диэпоксибутан и митомицин С. Слияние клеток анемии Фанкони с нормальными клетками или с клетками некоторых неродственных пациентов с анемии Фанкони оказывает корректирующее воздействие на ломкость хромосом, этот процесс называется комплементацией.
Этот процесс часто использовался в прошлом для скрининга мутировавшего гена пациента, до того, как стали широко доступны генные панели нового поколения, включающие известные гены анемии Фанкони.
Классический фенотип анемии Фанкони, четко определяющий ассоциированные с анемией Фанкони гены (FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BACH1/BRIP1, FANCL, FANCM, FANCN/PALB2, FANCP/SLX4, FANCQ/ERCC4, UBE2T, REV7), включает триаду, состоящую из недостаточности костного мозга, врожденных аномалий и повышенной ломкости хромосом. Эти гены могут мутировать у пациентов, имеющих один или все компоненты триады. Гены, которые, как было обнаружено, ассоциировались с одним или двумя, но не со всеми тремя компонентами, являются генами заболеваний, подобных анемии Фанкони (FANCO/RAD51C, RAD51, FANCS/ BRCA1, FANCR/EXCC2).
На долю гена FANCA приходится 64% случаев анемии Фанкони, на долю FANCC — 14%, а на долю FANG — 9%. FANCB, FANCD1/BRCA2, FANCD2, FANCE и FANCF в совокупности мутируют почти у 13% пациентов с анемией Фанкони. Остальные гены мутируют в редких случаях.
Белки, кодируемые генами FANC дикого типа, участвуют в распознавании повреждений ДНК и биохимическом пути репарации. Т.о., мутантные белки приводят к нестабильности генома и хрупкости хромосом. Белки FANC участвуют в др. клеточных процессах, таких как детоксикация активных форм кислорода, энергетический метаболизм и сигнализация цитокинов. Т.о., мутации FANC, вероятно, влияют на несколько клеточных и биохимических функций соответствующих белков, что в конечном итоге приводит к фенотипу анемии Фанкони. Наблюдаемая сложность и гетерогенность заболевания, вероятно, вызвана вовлечением множества клеточных и биохимических путей как у неродственных индивидов, так и у членов семьи с одной и той же генетической мутацией.
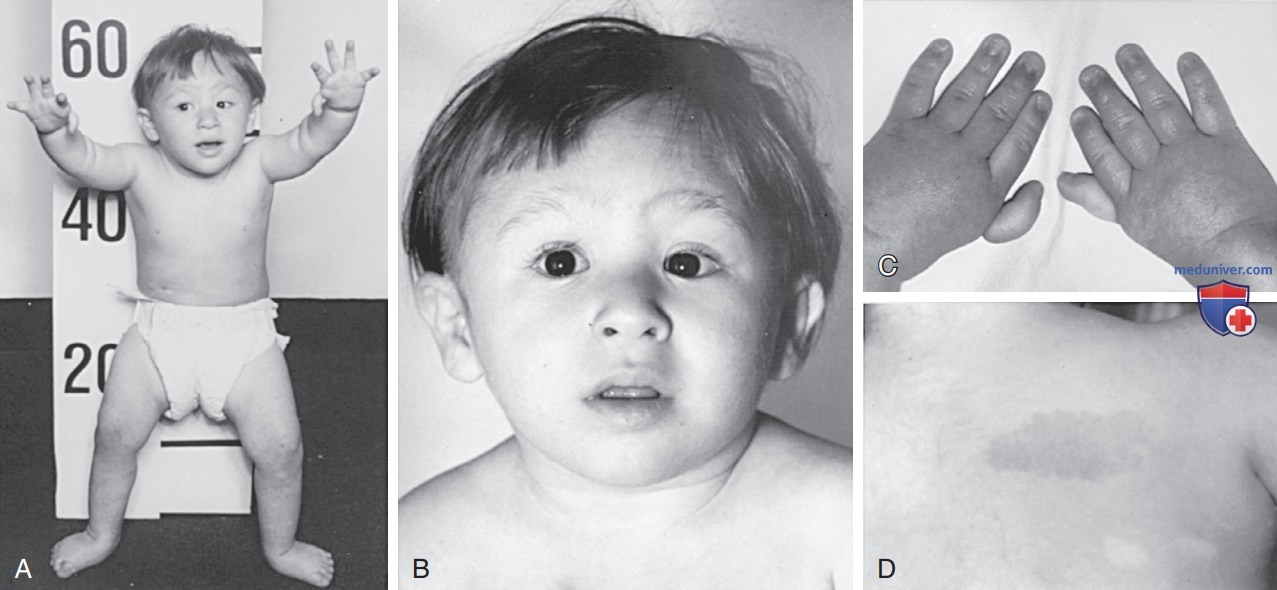
3. Клинические проявления. Наиболее распространенные врожденные пороки развития при анемии Фанкони являются скелетными и включают в себя отсутствие лучевых костей и/или затрагивают большие пальцы, которые м.б. гипопластичными, избыточными, раздвоенными или отсутствовать. Также могут наблюдаться пороки развития стоп, врожденный вывих бедра и пороки развития нижних конечностей (рис. 1 и табл. 2). Гиперпигментация кожи туловища, шеи и областей кожных складок, «кофейные пятна» на коже и витилиго, отдельно или в сочетании, встречаются с одинаковой частотой. Часто встречается низкий рост, который у некоторых пациентов усугубляется субнормальной секрецией СТГ или гипотиреозом.
Рисунок 1. 3-летний мальчик с анемией Фанкони с проявлением нескольких классических фенотипических особенностей заболевания: A — вид спереди; B — лицо; C — руки; D — правое плечо, вид сзади. Характерные особенности, которые следует отметить, включают в себя низкий рост, вывих бедер, микроцефалию, широкое основание носа, эпикантальные складки, микрогнатию, дефекты больших пальцев рук, «кофейные пятна» на коже с гипопигментированными областями внизу
Пациенты мужского пола с анемией Фанкони могут иметь недоразвитый пенис, неопущенные, атрофические или отсутствующие яички, а также гипоспадию или фимоз, и все такие пациенты бесплодны. У пациентов женского пола могут наблюдаться пороки развития влагалища, матки и яичников, фертильность у всех таких пациенток понижена. У многих пациентов наблюдаются характерные лицевые дисморфизмы, включая микроцефалию, маленькие глаза, эпикантальные складки, а также аномальная форма, размер или расположение ушей (см. рис. 1). Почки м.б. эктопическими, тазовыми, подковообразными, гипопластическими, диспластическими или отсутствовать. Также встречаются пороки развития ССС и ЖКТ.
У 10% пациентов с анемией Фанкони наблюдается задержка когнитивного развития. У новорожденных с анемией Фанкони обычно наблюдается ЗВУР и НМТР, они могут иметь пороки развития, соответствующие ассоциации VACTERL*/VACTERL-H** (пороки развития позвоночника, анальная атрезия, пороки развития сердца, трахеопищеводный свищ с атрезией пищевода, структурные аномалии почек и конечностей с гидроцефалией).
P.S. ** VACTERL-H (VACTERL with Hydrocephalus) — синдром VACTERL с гидроцефалией.
Недостаточность костного мозга обычно появляется в течение первого десятилетия жизни. Тромбоцитопения, макроцитоз эритроцитов и повышенный уровень фетального Hb (HbF*) в результате стресса костного мозга часто проявляются первыми.
На этих стадиях аспират и биопсия костного мозга часто показывают гипопластический образец. Впоследствии у больных развивается нейтропения, а затем анемия. В большинстве случаев, обычно через несколько лет, развивается тяжелая аплазия.
4. Осложнения. В дополнение к низким показателям крови и физическим патологиям, пациенты с анемией Фанкони входят в группу высокого риска по развитию онкологических заболеваний. Наиболее часто встречающимися солидными опухолями являются сквамозно-клеточная карцинома головы и шеи (риск в 600 раз выше, чем в общей популяции), а также карцинома верхнего отдела пищевода (риск в 2000 раз выше), вульвы (риск в 3000 раз выше) и/или ануса, шейки матки и нижнего отдела пищевода. Манифестация злокачественной солидной опухоли наблюдается гораздо раньше, чем в общей популяции, причем средний возраст начала сквамозно-клеточной карциномы в популяции с анемией Фанкони составляет 33 года по сравнению с 60-70 годами в общей популяции. В патогенезе сквамозно-клеточной карциномы подозревают ВПЧ.
Могут развиваться доброкачественные и злокачественные опухоли печени (аденомы, гепатомы), которые обычно связаны с андрогенной терапией при апластической анемии.
Андрогены также вовлечены в этиологию печеночной пурпуры (наполненных кровью печеночных синусоидов), которая является обратимой, когда терапия андрогенами прекращается. При анемии Фанкони часто встречаются клональные цитогенетические аномалии костного мозга, которые при последующем наблюдении м.б. либо стабильными, периодически выявляемыми, либо прогрессирующими. Кумулятивная частота клональной и злокачественной миелоидной трансформации к 18 годам, включающая клональные цитогенетические аномалии костного мозга, миелодиспластический синдром и острый миелоидный лейкоз, составляет 75%. Исследование 2003 г. показало, что к 40 годам совокупная заболеваемость лейкозом составляет 33%.
5. Диагностика. Анемию Фанкони следует подозревать у всех детей и молодых людей с необъяснимыми цитопениями. Аномальные гематологические показатели и характерные физические патологии предполагают диагноз, который м.б. подтвержден исследованием хромосомных разрывов в лимфоцитах, выполненным с добавлением и без добавления сшивающих агентов, таких как диэпоксибутан и митомицин С. На повышенную ломкость хромосом указывают спонтанно возникающие хроматидные разрывы, перестройки, бреши, эндоредупликации и хроматидные обмены в лимфоцитах крови, культивируемых с фитогемагглютинином, а также в культивируемых фибробластах кожи, что подчеркивает конституциональную природу.
С добавлением диэпоксибутана и митомицина С ломкость удивительным образом усиливается в культурах лимфоцитов пациентов с анемией Фанкони по сравнению с контрольными культурами. Анализ аномальных хромосом и генетическое тестирование в рамках пренатальной диагностики м.б. выполнены на клетках амниотической жидкости или на ткани из биопсии ворсин хориона. Согласно исследованию хромосомных разрывов, ни один из др. типов врожденной панцитопении не ассоциирован с выраженной гиперчувствительностью in vitro к диэпоксибутану и митомицину С. У 10-15% пациентов с подозрением на анемию Фанкони наблюдается соматический мозаицизм, но может отсутствовать характерно высокая степень ломкости хромосом в лимфоцитах, что указывает на наличие смешанных популяций соматических клеток, некоторые из которых имеют 2 аномальных аллеля, а некоторые только 1.
Последняя из двух популяций лимфоцитов берет начало от части гемопоэтических стволовых клеток, которые подверглись спонтанной генной коррекции соматических клеток по одному аллелю. Тестирование фибробластов кожи следует проводить, если подозрение на анемию Фанкони является высоким, несмотря на «-» результаты тестирования лимфоцитов периферической крови.
{5С} Для подтверждения или исключения анемии Фанкони используют ДЭБ-тест — проба с диэпоксибутаном*.
P.S. * КР РФ Миелодиспластический синдром, 2020 г. и КР РФ Апластическая анемия, 2021 г..
Из-за большого количества генов FANC генетическая диагностика традиционно начиналась с тестирования комплементации, определяя, снижается ли или устраняется клеточная гиперчувствительность к сшивающим агентам после генерации гибрида клеток пациента с известными генетически комплементными клетками или после трансдукции клеток пациента известным геном FANC дикого типа. Мутантный аллель разбавляется, когда ген FANC дикого типа той же группы комплементации вводится с помощью вышеописанных методов, что приводит к коррекции аномальной хрупкости хромосом, первоначально наблюдаемой у пациента. Секвенирование нового поколения практически отменило необходимость двухэтапного генетического тестирования (тестирование на группу комплементации с последующим тестированием специфических генов) и используется чаще всего.
Секвенирование нового поколения является эффективным и точным методом диагностики анемии Фанкони, но иногда м.б. ограничено трудностями интерпретации неизвестных вариантов. Когда не получается выявить конкретные варианты, являющиеся причиной болезни, можно использовать методы анализа вариаций числа копий и их локализацию с высоким разрешением, а затем провести поиск новых ассоциированных генов по всему геному.
После подтверждения диагноза анемия Фанкони необходим всесторонний скрининг на наличие потенциальных нарушений состояния здоровья. Использование ионизирующего излучения при визуализации должно быть сведено к минимуму насколько это возможно из-за канцерогенного риска, присущего этому заболеванию с генетической нестабильностью; КТ должна быть заменена МРТ, когда это возможно. В дополнение к детальному изучению истории перенесенных заболеваний и тщательному физикальному обследованию, скрининг должен включать УЗИ БП и ЭхоКГ, чтобы исключить внутренние врожденные пороки развития. Др. методы визуализации м.б. применены по мере необходимости и на основе первоначального скрининга.
В это время м.б. организованы специальные консультации по выявленным патологиям и нарушениям здоровья, ведущим к недееспособности. Если скорость роста ребенка ниже, чем ожидается, то для оценки дефицита СТГ необходимо обследование эндокринной системы. Анализ крови должен включать оценку состояния почек, печени, ЩЖ, метаболической и иммунной систем.
{5С} Всем детям, а также пациентам с семейным анамнезом гематологической патологии (лейкемии, тромбоцитопении, апластические анемии) и с дебютом с нетяжелой, медленно прогрессирующей цитопении рекомендуется молекулярно-цитогенетическое исследование (FISH-метод**) на одну пару хромосом на анемию Фанкони для ДД с врожденными формами костномозговой недостаточности***.
P.S. ** Флуоресцентная гибридизация in situ.
P.S. *** КР РФ Апластическая анемия, 2021 г.
6. Лечение. Пациентов с анемией Фанкони должен вести врач-гематолог, предпочтительно тот, который специализируется на синдромах врожденной недостаточности костного мозга, вместе с многопрофильной группой специалистов, когда это возможно. При постановке диагноза необходимо провести детальную оценку показателей крови пациента, функции костного мозга, роста, развития, а также функций др. органов. При редких расстройствах, таких как анемия Фанкони, необходимы специальные методы исследования для оценки экономической эффективности инструментов ведения болезни. Однако рекомендуются и широко применяются нижеследующие тесты.
Если гематологические патологии имеют легкую или умеренную степень тяжести и являются стабильными и нет необходимости в гемотрансфузии, пациенты м.б. обследованы с помощью анализа периферической крови каждые 3 мес и ежегодного исследования костного мозга методом аспирации для выявления клональных цитогенетических аномалий, миелодиспластическо синдрома и острого миелоидного лейкоза. Во время исследования костного мозга также может периодически проводиться биопсия костного мозга для оценки изменений в процентном выражении насыщенности костного мозга клетками и фиброза. Мониторинг можно проводить чаще при необходимости, напр., когда происходит снижение показателей крови. Уровень глюкозы следует измерять ежегодно или раз в два года, в зависимости от степени гипергликемии, обнаруженной при первоначальном тестировании. Скрининг на гипотиреоз следует проводить ежегодно.
Пациенты должны обследоваться на наличие солидных опухолей не реже одного раза в год с тщательным физикальным осмотром, включающим всестороннее обследование кожи, полости рта и др. органов на наличие необычных новообразований. После определенного возраста (напр., 10 лет) или после трансплантации гемопоэтических стволовых клеток также рекомендуется флюороскопическое исследование ротоглотки и анализ на скрытую кровь в кале. С наступлением менструации пациентки женского пола должны ежегодно проходить обследование на гинекологический рак. Рекомендуется введение четырехвалентной вакцины против ВПЧ для снижения риска развития сквамозно-клеточной карциномы.
Тестирование на ломкость хромосом (и/или специфическое генетическое тестирование) должно быть предложено родителям пациентов, пораженных этим заболеванием, а также их братьям и сестрам, для идентификации др. пораженных лиц. Типирование HLA пациента, биологических родителей и родственных братьев и сестер для будущей трансплантации гемопоэтических стволовых клеток также должно происходить в самом начале.
{5С} Всем пациентам при диагностике апластической анемии, а также их сиблингам рекомендуется HLA ДНК-типирование по низкому разрешению по 6 аллелям локусов А,В и DRB1 с консультацией в трансплантационном центре с целью выбора метода лечения и поиска потенциального донора костного мозга*.
P.S. * КР РФ Апластическая анемия, 2021 г..
Трансплантация гемопоэтических стволовых клеток — это единственное радикальное лечение гематологических нарушений, наблюдаемых у пациентов с анемией Фанкони. За последние 2 десятилетия результаты трансплантации гемопоэтических стволовых клеток значительно улучшились благодаря модифицированным режимам со сниженной интенсивностью, которые понизили токсичность, которой подвергаются пациенты с анемией Фанкони, имеющие чувствительность к агентам, повреждающим ДНК, таким как алкилирующие препараты и облучение. Те пациенты, которые проходят трансплантацию с использованием HLA-совместимого родственного донора без облучения на предварительном этапе, имеют показатели общей 3-5-летней выживаемости >80%. Общая выживаемость пациентов с анемией Фанкони, трансплантированных от полностью совместимого неродственного донора, составляет 65-70%.
У пациентов, которым трансплантация была проведена до того, как они получили множественные гемотрансфузии или развили клональную и злокачественную миелоидную трансформацию (миелодиспластический синдром или острый миелоидный лейкоз), показатели выживаемости лучше. Показатели выживаемости выше у пациентов, перенесших трансплантацию в возрасте <10 лет.
Усовершенствование метода HLA-типирования с высоким разрешением привело к более оптимальному отбору неродственных доноров и улучшению результатов. Развитие молекулярных технологий привело к появлению предим-плантационной генетической диагностики родительских бластомеров, что позволило имплантировать непораженные бластомеры и в результате создать HLA-совместимого родственного донора без анемии Фанкони.
Андрогены вызывают ответную реакцию у ~70% пациентов, о чем свидетельствует ретикулоцитоз и повышение уровня Hb в течение 1-2 мес. Затем может увеличиться количество лейкоцитов, а затем и количество тромбоцитов. После проявления первоначального ответа на терапию показатели крови могут продолжать улучшаться в течение многих месяцев, пока не будет достигнут максимальный ответ. Если терапия начинается с низкой дозы, то дозу андрогена можно увеличивать каждые 3-4 нед до тех пор, пока не будет замечено никаких серьезных побочных эффектов и пока не будет достигнут желаемый ответ. Если терапия начинается с высокой дозы андрогена, то доза м.б. медленно снижена до минимальной дозы, достаточной для поддержания показателей крови на необходимом уровне.
Чаще всего из андрогенных препаратов используются пероральные оксиметалон и даназол. Пациенты обычно перестают реагировать на андрогены через несколько месяцев или лет, по мере прогрессирования недостаточности костного мозга или развития миелодиспластического синдрома / острого миелоидного лейкоза. Т.о., лечение андрогенами не является радикальным, а используется скорее в качестве переходной терапии в ожидании подходящего донора для трансплантации гемопоэтических стволовых клеток или в период оценки рисков и пользы трансплантации. Побочные эффекты андрогенов включают маскулинизацию, усиление линейного роста, учащение перепадов настроения или повышение агрессивности, повышение уровня печеночных ферментов, застой желчи, печеночную пурпуру и опухоли печени. Скрининг на выявление таких побочных эффектов следует проводить регулярно.
Потенциал терапии анемии Фанкони с помощью рекомбинантного фактора роста (цитокина) не определен. Гранулоцитарный колониестимулирующий фактор обычно может индуцировать повышение абсолютного количества нейтрофилов; однако может существовать повышенный риск экспансии клеток костного мозга с клональными цитогенетическими аномалиями, такими как моносомия 7-й хромосомы. В одном исследовании комбинированная терапия, состоящая из гранулоцитарного колониестимулирующего фактора, вводимого п/к ежедневно или каждые 2 дня вместе с эритропоэтином, вводимым п/к или в/в 3 р/нед, привела к улучшению показателей нейтрофилов у большинства пациентов и устойчивому повышению уровня Hb и тромбоцитов у ~1/3 пациентов. Большинство пациентов перестали отвечать на терапию из-за прогрессирования заболевания костного мозга.
{5С} В посттрансплантационном периоде всем детям с апластической анемией не рекомендуется плановое использование гранулоцитарного колониестимулирующего фактора.
{5С} Всем детям с апластической анемией во время проведения курса иммуносупрессивной терапии не рекомендуется назначение гранулоцитарного колониестимулирующего фактора.
{5С} Пациентам с апластической анемией при наличии показаний рекомендуется проведение заместительной трансфузионной терапии компонентами крови.
{5С} Пациентам с апластической анемией и посттрансфузионной перегрузкой железом рекомендуется применение хелаторной терапии деферазироксом (противопоказан пациентам в возрасте до 2 лет) в начальной дозе 10 мг/кг в сутки, с дальнейшим постепенным увеличением дозы до 30 мг/кг в сутки при отсутствии признаков токсичности препарата*.
P.S. * КР РФ Апластическая анемия, 2021 г.
7. Прогноз. Развитие методов поддерживающей терапии, тщательное отслеживание известных осложнений, оперативное вмешательство и усовершенствованные методы трансплантации привели к тому, что пациенты с анемией Фанкони доживают до 30 и более лет. К сожалению, существует повышенный риск развития солидных опухолей после трансплантации гемопоэтических стволовых клеток. Напр., риск развития рака органов головы и шеи увеличивается в 4,4 раза и ускоряется на ~15 лет по сравнению с пациентами, не прошедшими трансплантацию. Кумулятивная частота возникновения ЗНО через 20 лет после трансплантации составляет 35-40%. Некоторые повышенные риски м.б. связаны с использованием ДНК-повреждающих агентов или возникновением РТПХ.
б) Синдром Швахмана-Даймонда:
1. Этиология и эпидемиология. Синдром Швахмана-Даймонда наследуется АуР-путем и встречается во всех расовых и этнических группах. Как и в случае с анемией Фанкони, синдром Швахмана-Даймонда — это мультисистемное расстройство. Однако негематологические проявления синдрома Швахмана-Даймонда существенно различны и обычно включают экзокринную недостаточность ПЖЖ и патологии скелета, такие как метафизарная дисплазия (табл. 3). Синдром Швахмана-Даймонда представляет собой рибосомопатию, и основной дефект затрагивает сборку рибосом. Не наблюдается повышенного количества разрывов хромосом после проведения теста с диэпоксибутаном на ломкость хромосом в лимфоцитах, пораженных синдромом Швахмана-Даймонда.
2. Патология. С синдромом Швахмана-Даймонда ассоциированы три гена. SBDS — это 1-й ген, который был описан в 2003 г. как мутировавший при синдроме Швахмана-Даймонда. SBDS расположен на участке 7ql 1 7-й хромосомы, мутация этого гена встречается в 80-90% случаев синдрома Швахмана-Даймонда. SBDS играет важную роль на поздней стадии созревания npe-60S рибосомальной субъединицы, связываясь с ГТФазой-фактором элонгации типа 1 (EFL1) и облегчая высвобождение eIF6 для формирования 80S моносом. DNAJC21 является 2-м зарегистрированным геном, мутировавшим при синдроме Швахмана-Даймонда. Функция гена человека DNAJC21, и его гомолога Jjjl в дрожжах Saccharomyces cerevisiae необходима для высвобождения и рециркуляции гетеродимера Arxl/Albl из факторов биогенеза субъединицы пре-60S. 3-й обнаруженный ген, мутировавший при синдроме Швахмана-Даймонда, — это ген EFL1.
Генетические дефекты, лежащие в основе синдрома Швахмана-Даймонда, указывают на то, что последний этап биогенеза рибосом ассоциирован с панцитопенией (чаще всего с нейтропенией) и гипопластическим костным мозгом. Дефекты в рибосомных белках, которые задействованы на более ранних стадиях созревания субъединиц рибосомы и которые являются структурными компонентами рибосомы, связаны преимущественно с анемией и парциальной красноклеточной аплазией.
Недостаточность ПЖЖ обусловлена недостаточностью развития ацинарных клеток ПЖЖ при синдроме Швахмана-Даймонда, при этом наблюдается выраженное жировое перерождение тканей ПЖЖ. Недостаточность костного мозга характеризуется дисфункциональными гемопоэтическими стволовыми клетками, ускоренным апоптозом предшественников костного мозга и дефектным микроокружением костного мозга, которое не поддерживает нормальный гемопоэз.
3. Клинические проявления. У большинства пациентов с синдромом Швахмана-Даймонда с рождения наблюдаются симптомы мальабсорбции жиров, что вызвано недостаточностью ПЖЖ, но при этом стеаторея не всегда выражена. У 50% пациентов с возрастом наблюдается улучшение секреции панкреатических ферментов. В клинической картине могут преобладать осложнения в виде анемии, нейтропении или тромбоцитопении. Могут возникать бактериальные и грибковые инфекции, вторичные по отношению к нейтропении, дисфункции нейтрофилов и иммунодефициту. Низкий рост является характерной особенностью синдрома Швахмана-Даймонда.
Большинство пациентов демонстрируют нормальную скорость роста, но при этом показатели роста и МТ постоянно находятся ниже 3-го процентиля. В редких случаях взрослые пациенты с синдромом Швахмана-Даймонда достигает 25-го процентиля по росту. Хотя патологии скелета разнообразны, классическая картина включает в себя метафизарную дисплазию, остеопению, замедленное появление вторичных ядер окостенения, короткие или расширенные ребра и дистрофию ГК. У некоторых пациентов наблюдается гепатомегалия и повышение уровня печеночных ферментов. У большинства пациентов наблюдаются аномалии зубов и плохое состояние полости рта. У многих пациентов возникают нейрокогнитивные проблемы и нарушения социальной коммуникации.
4. Результаты лабораторных исследований. Недостаточность ПЖЖ при синдроме Швахмана-Даймонда связана со снижением стандартизированных по возрасту уровней трипсиногена и панкреатической изоамилазы в сыворотке крови. Поскольку уровень панкреатической изоамилазы в сыворотке крови физиологически низкий в первые 3 года жизни, а снижение сывороточного трипсиногена обычно наблюдается у маленьких детей и повышается с возрастом, тестирование обоих ферментов м.б. информативно. Уровень панкреатической эластазы в кале у пациентов с синдромом Швахмана-Даймонда часто снижен. Наблюдается нарушение всасывания жирорастворимых витаминов (A, D, Е и К), и поэтому для оценки последствий нарушения всасывания жиров полезно измерение уровня витаминов A, D и Е, а также времени свертывания крови.
УЗИ или КТ позволяют визуализировать жировое перерождение ткани ПЖЖ. Мальабсорбция жиров м.б. подтверждена с помощью анализа кала с его сбором в течение 72 ч. Функциональное исследование ПЖЖ показывает значительное нарушение секреции ферментов, но с сохранением функции протоков. Такое исследование проводится редко, обычно оно заменяется анализом сыворотки крови и фекальных ферментов, а также визуализацией ПЖЖ.
Нейтропения наблюдается у ~70% пациентов с синдромом Швахмана-Даймонда при выявлении заболевания и почти у 100% пациентов при последующем наблюдении. Нейтропения носит хронический характер, но м.б. стойкой или перемежающейся, а также легкой, умеренной или тяжелой степени. Нейтропения может выявляться у некоторых новорожденных при сепсисе. Нейтрофилы могут иметь патологии подвижности, миграции и хемотаксиса из-за изменений функций цитоскелета или микротрубочек. Анемия, тромбоцитопения и панцитопения наблюдаются в 40-66, 40-60 и 21-44% случаев соответственно. Панцитопения м.б. тяжелой в результате резко выраженной апластической анемии.
Образцы биопсии и аспираты костного мозга показывают разл. степень гипоплазии и жировой инфильтрации костного мозга. Однако в раннем возрасте или если у пациентов развивается миелодиспластический синдром или лейкоз, костный мозг м.б. нормоцеллюлярным или даже гиперцеллю-лярным. У пациентов также могут наблюдаться дефекты В-клеток с одним или более из следующих признаков: низкий уровень IgG или его подклассов, низкий процент циркулирующих В-лимфоцитов, снижение пролиферации В-клеток in vitro и отсутствие выработки специфических АТл. Пациенты могут иметь низкий процент циркулирующих Т-клеток, подклассов или естественных клеток-киллеров и сниженную пролиферацию Т-клеток in vitro.
5. Диагностика. Клинический диагноз синдром Швахмана-Даймонда основывается на наличии признаков дисфункции костного мозга и экзокринной недостаточности ПЖЖ. Однако до 20% пациентов не имеют четких свидетельств наличия экзокринных дефектов ПЖЖ при постановке диагноза. Поэтому рекомендуется, чтобы для всех пациентов с гипопластическим/апластическим костным мозгом неизвестной этиологии была рассмотрена возможность проведения генетического тестирования на синдром Швахмана-Даймонда. Анализ на мутации генов SBDS, DNAJC21 и EFL1 имеет решающее значение для подтверждения диагноза во всех или почти во всех случаях синдрома Швахмана-Даймонда.
Синдром Пирсона, включающий рефрактерную сидеробластную анемию, цитоплазматическую вакуолизацию предшественников костного мозга, метаболический ацидоз, экзокринную недостаточность ПЖЖ и диагностические мутации митохондриальной ДНК, похож на синдром Швахмана-Даймонда, но отличается от него клиническим течением, морфологическими особенностями костного мозга и иными мутациями генов. Кроме того, при синдроме Пирсона с рождения до 1 года наблюдается не нейтропения, а анемия тяжелой степени, требующая гемотрансфузии.
Синдром Швахмана-Даймонда имеет некоторые общие характерные признаки с анемией Фанкони, такие как дисфункция костного мозга и отставание в росте, но синдром Швахмана-Даймонда легко дифференцируется от анемии Фанкони по недостаточности ПЖЖ с нарушением всасывания жиров, жировых перерождений тканей ПЖЖ, подтверждаемых с помощью диагностической визуализации, характерных скелетных патологий, не наблюдаемых при анемии Фанкони, а также соответствующих норме результатов теста на ломкость хромосом с диэпоксибутаном и митомицином С.
Отличить синдром Швахмана-Даймонда от врожденного дискератоза на основании только клинических данных и уровня панкреатических ферментов м.б. невозможно, а измерение длины теломер может способствовать постановке правильного диагноза.
В сложных случаях синдрома врожденной недостаточности костного мозга, которые не м.б. легко классифицированы, в постановке диагноза может помочь расширенный генетический анализ с применением секвенирования нового поколения панели всех известных генов, ответственных за синдром врожденной недостаточности костного мозга, или объективное тестирование методом секвенирования всего экзома/генома.
6. Осложнения. Пациенты с синдромом Швахмана-Даймонда предрасположены к миелодиспластическому синдрому и лейкозной трансформации. У -25% пациентов к 18 годам развиваются клональные цитогенетические аномалии костного мозга, миелодиспластический синдром или лейкоз. У 1/3 пациентов лейкоз развивается к 30 годам. Особенно часто встречается изохромосома 7q[i(7q)]. Это позволяет предположить, что она является довольно специфичным клональным маркером синдрома Швахмана-Даймонда и м.б. связана с присутствием мутантных генов SBDS на 7ql 1. Др. клональные хромосомные аномалии включают моносомию 7, i(7q) в сочетании с моносомией 7, делеции или транслокации, включающие часть 7q, и делеции 20q [Del(20q)].
I(7q) и del(20q) ассоциированы с относительно низким риском и очень медленным развитием миелодиспластический синдром или лейкозной трансформации; однако их прогностическое значение и значение др. клональных изменений костного мозга требуют дальнейшего проспективного мониторинга.
7. Лечение. Мальабсорбция жиров отвечает на пероральное восполнение дефицита панкреатических ферментов и добавление в рацион жирорастворимых витаминов, вводимых в соответствии с рекомендациями, аналогичными тем, которые применяются при муковисцидозе. Следует разработать долгосрочный план мониторинга изменений показателей периферической крови, требующих корректирующих действий, и поиска ранних признаков злокачественной миелоидной трансформации. Последнее требует периодического исследования аспирата костного мозга, цитогенетического анализа и биопсии костного мозга. Одна из рекомендаций состоит в том, чтобы проводить исследование костного мозга каждые 1-3 года и ОАК каждые 3 мес.
Ежедневное п/к введение гранулоцитарного колониестимулирующего фактора при глубокой нейтропении эффективно для достижения устойчивого повышения количества нейтрофилов. Некоторые пациенты нуждаются в трансфузионной поддержке для лечения тяжелой анемии или тромбоцитопении. Опыт применения эритропоэтина ограничен. У некоторых пациентов, получавших [андрогены + стероиды], показатели крови улучшились.
Единственным методом радикального лечения тяжелой недостаточности костного мозга и прогрессирующего миелодиспластический синдром или лейкоза при синдроме Швахмана-Даймонда является аллогенная трансплантация гемопоэтических стволовых клеток, хотя опыт ее применения ограничен. Смертность, связанная с традиционной миелоаблативной трансплантацией гемопоэтических стволовых клеток, составляет 35-50% пациентов. Режимы кондиционирования сниженной интенсивности, включающие флударабин, по-видимому, более безопасны и эффективны при трансплантации гемопоэтических стволовых клеток у пациентов с синдромом Швахмана-Даймонда.
Лечение прогрессирующего миелодиспластического синдрома и острого миелоидного лейкоза, как правило, малоэффективно, а исход часто неблагоприятный.
8. Прогноз. Точная продолжительность жизни пациентов с синдромом Швахмана-Даймонда неизвестна; анализ опубликованных случаев показал, что медиана выживаемости составляет 35 лет. Поскольку число недиагностированных пациентов с легкой или бессимптомной формой заболевания неизвестно, общий прогноз м.б. лучше, чем считался ранее. У 50% пациентов наблюдается спонтанная коррекция недостаточности ПЖЖ в результате улучшения секреции панкреатических ферментов.
Хотя у всех пациентов при постановке диагноза наблюдается некоторая степень гематологической цитопении, изменения у большинства из них носят легкий или умеренный характер и не требуют терапевтического вмешательства. Тяжелая нейтропения хорошо реагирует на гранулоцитарный колониестимулирующий фактор, но есть опасения, что гранулоцитарный колониестимулирующий фактор может способствовать росту эволюционирующих клонов миелодиспластический синдром или лейкоза из-за мощного эффекта стимулирования роста клеток костного мозга. Трансплантация гемопоэтических стволовых клеток при тяжелой недостаточности костного мозга дает 50-70% выживаемости, но в настоящее время разрабатываются и более безопасные протоколы лечения. Все еще представляет опасность злокачественная трансформация костного мозга.
в) Врожденный дискератоз:
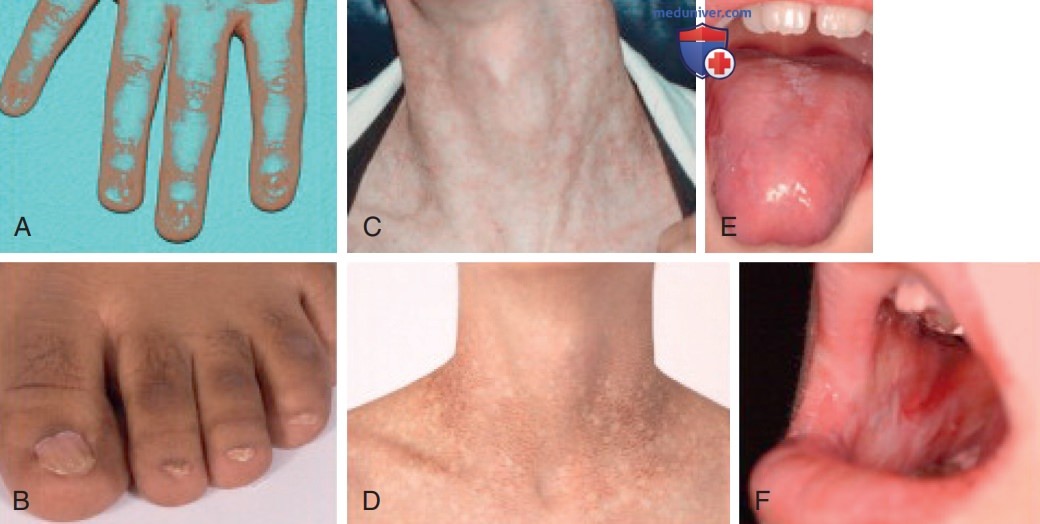
1. Этиология и эпидемиология. Врожденный дискератоз — это наследственное мультисистемное заболевание, связанное с теломерами хромосом. При первом описании заболевания была предложена диагностическая триада кожно-слизистых симптомов, которая включала в себя диспластические ногти, нарушения пигментации кожи в виде кружевного, сетчатого рисунка на шее и/или в верхней части ГК, а также лейкоплакию полости рта (рис. 2). Однако такая триада может наблюдаться не у всех пациентов. При возникновении симптомов диагностической триады кожные и ногтевые симптомы обычно проявляются в первые 10 лет жизни, тогда как лейкоплакия полости рта м.б. замечена позже. Эти проявления имеют тенденцию прогрессировать с возрастом пациентов. Недостаточность костного мозга разл. степени тяжести наблюдаются у 90% пациентов.
Рисунок 2. Диагностическая триада характерных признаков врожденного дискератоза: A, B — дистрофические ногти на руках и ногах; C, D — нарушения пигментации кожи в виде кружевного, сетчатого рисунка на шее и в верхней части грудной клетки; E, F — лейкоплакия полости рта в области языка и слизистой оболочке щек
Тяжелая апластическая анемия встречается в 50% случаев, причем возраст начала заболевания варьируется в зависимости от группы генетической мутации. В некоторых группах заболевание обычно начинается на первом десятилетии жизни (напр., при мутациях в генах DKC1, TINF2, PARN), тогда как в др. группах оно обычно начинается после 1-го десятилетия (напр., при мутациях в генах TERT, TERC). Помимо прогрессирующей недостаточности костного мозга, пациенты с врожденным дискератозом также подвержены высокому риску развития фиброза легких и печени, др. врожденных аномалий, а также предрасположенности к солидным опухолям и миелодиспластический синдром или острый миелоидный лейкоз. Врожденный дискератоз встречается редко, причем частота заболеваемости в детском возрасте составляет 4:1 000 000 населения в год.
2. Патология. Врожденный дискератоз генетически гетерогенен, и у пациентов наблюдаются мутации в генах, кодирующих компоненты теломеразного комплекса (TERT, DKC1, TERC, NOP10 и NHP2), белок разборки Т-петли (RTEL1), кэппинговый комплекс теломер (СТС1, STN1), теломершелтериновый комплекс (TINF2, ACD), теломераз-траффинговый белок (TCAB1/WRAP53), деаденилазу поли(А)-специфической рибонуклеазы (PARN), а также мутации в недавно выявленном гене с пока еще неясной ролью (NAFF).
Все компоненты имеют решающее значение для поддержания длины теломер. Х-сцепленная рецессивная форма врожденного дискератоза соответствует Xq28, а в гене DKC1, который кодирует ядерный белок диске-рин, были выявлены многие мутации. АуД-форма заболевания обусловлена мутациями в генах TINF2, TERC, TERT, RTEL1, ACD и NAF1. АуР-форма врожденного дискератоза связана с мутациями в генах NOP10, NHP2, PARN, TCAB1/WRAP53, СТС1 и STN1, а также в генах TERT, RTEL и ACD. Из-за нарушения механизма поддержания длины теломер при всех 3 наследственных формах врожденного дискератоза в клетках периферической крови всех пациентов наблюдаются чрезвычайно короткие теломеры (<1-го процентиля по возрасту).
Выявление чрезвычайно коротких теломер в лимфоцитах с помощью метода автоматизированной многоцветной флуоресцентной гибридизации in situ (метод FISH), имеет чувствительность 97% и специфичность 91% для врожденного дискератоза. У 70% пациентов, у которых клинические проявления соответствуют ДК врожденного дискератоза, наблюдается патогенный вариант в 1 из известных генов, связанных с врожденным дискератозом. Наиболее распространены мутации в гене DKC1 (20-25% пациентов), за ними следуют мутации в генах TINF2 (12-20% пациентов), TERC (5-10% пациентов), RTEL1, TERT и СТС1. Остальные генетические мутации были описаны в 6 или менее семьях.
Недостаточность костного мозга, вероятно, вызвана прогрессирующим истощением и деплецией гемопоэтических стволовых клеток из-за преждевременного старения, апоптоза или нестабильности хромосом, которые проявляются в виде панцитопении.
3. Клинические проявления. Клинические критерии классического варианта врожденного дискератоза были впервые описаны в 2006 г. и включают наличие по крайней мере 2 из 4 основных признаков — аномальной пигментации кожи, дистрофии ногтей, лейкоплакии и недостаточности костного мозга, а также 2 или более др. соматических признаков, ассоциированных с врожденным дискератозом. Однако постановка диагноза по-прежнему остается сложной задачей, поскольку у отдельных людей клинические признаки врожденного дискератоза проявляются с разной скоростью и в разном возрасте, даже у пациентов в пределах одной семьи. У 30% людей с врожденным дискератозом патогенные мутации в известных генах, связанных с врожденным дискератозом, не м.б. идентифицированы.
Клиническая картина варьируется от пациентов, у которых сначала развивается недостаточность костного мозга, а затем, спустя годы, проявляются др. классические симптомы, такие как аномалии ногтей, до пациентов, у которых наблюдаются серьезные патологии ногтей и нарушения пигментации кожи при проявлении заболевания, но сохраняется нормальная функция костного мозга. При классическом течении заболевания пигментация кожи и изменения ногтей обычно появляются первыми, обычно на первом десятилетии жизни. Недостаточность костного мозга обычно развивается в течение первых двух десятилетий жизни, причем у 80% пациентов недостаточность костного мозга развивается к возрасту 30 лет, но рано или поздно недостаточность костного мозга возникает почти у 90% пациентов.
«Кружевная», сетчатая пигментация кожи, поражающая лицо, шею, грудь и руки, является распространенным явлением (89%). Степень пигментации увеличивается с возрастом и может затрагивать всю поверхность кожи. Также может наблюдаться телеангиэктатический эритематозный компонент. Вторым по распространенности симптомом является дистрофия ногтей обеих рук и ног (88%). Она обычно начинается с образования продольных борозд, расслоения или образования птеригиума и может прогрессировать до полной потери ногтей. Лейкоплакия обычно поражает слизистую оболочку полости рта (78%), особенно язык, но может также наблюдаться на конъюнктиве и на анальной, уретральной или генитальной слизистых оболочках. Часто встречается чрезмерное слезотечение (эпифора), вторичное по отношению к непроходимости носослезных протоков, оно наблюдается у 30% пациентов.
У 25% пациентов наблюдаются трудности в обучении и/или задержка развития. Гипергидроз ладоней и подошв, выпадение и седина волос, кариес или выпадение зубов, стриктура пищевода, легочные заболевания со сниженной диффузионной способностью и/или рестриктивный дефект вследствие легочного фиброза, а также аномалии легочной и сосудистой системы и низкий рост наблюдаются у 15-20% пациентов.
К глазным аномалиям относятся конъюнктивит, блефарит, выпадение ресниц, косоглазие, катаракта и атрофия зрительного нерва. Патологии скелета включают остеопороз, аваскулярный некроз головки бедра или плечевой кости, аномальную трабекуляцию костей, сколиоз и гипоплазию нижней челюсти. Патологии МПС включают гипопластические яички, гипоспадию, фимоз, стеноз уретры и подковообразную почку. Нарушения со стороны ЖКТ, такие как сосудистые поражения, вызывающие кровотечение, гепатомегалия, пептические язвы и фиброз, наблюдаются в 10% случаев.
4. Результаты лабораторных исследований. Первичным гематологическим изменением при врожденном дискератозе обычно является или тромбоцитопения, или анемия, или и то, и другое, а затем следуют панцитопения и апластическая анемия. Эритроциты часто макроцитарные, а уровень HbF повышен. Исходные образцы костного мозга м.б. нормоцеллюлярными или гиперцеллюлярными, но со временем происходит симметричное истощение всех кроветворных линий. У некоторых пациентов наблюдаются иммунологические нарушения, включая снижение или повышение уровня Ig, снижение количества В- и/или Т-лимфоцитов, а также снижение или отсутствие пролиферативной активности лимфоцитов в присутствии фитогемагглютинина. Такие нарушения особенно часто встречаются в тяжелой форме при мутациях в гене DKC1.
В отличие от пациентов с анемией Фанкони, пациенты с врожденным дискератозом не имеют повышенного количества хромосомных разрывов в стимулированных фитогемагглютинином лимфоцитах спонтанно или после воздействия сшивающих агентов. Однако первичные культуры фибробластов кожи имеют аномальные морфологические особенности и аномальную скорость удвоения популяции и демонстрируют многочисленные несбалансированные хромосомные перестройки, такие как дицентрические или трицентрические, а также транслокации в отсутствие диэпоксибутана и митомицина С. Эти проявления свидетельствуют о дефекте, который предрасполагает клетки пациента к хромосомным перестройкам и, возможно, к повреждению ДНК.
5. Диагностика. Следующие аномалии наблюдаются у пациентов с врожденным дискератозом, но отсутствуют у пациентов с анемией Фанкони: дистрофия ногтей, лейкоплакия, аномалии зубов, гипергидроз ладоней и подошв, выпадение волос. Существует несколько относительно более тяжелых форм врожденного дискератоза. Синдром Хойераала-Хрейдарссона — это мультисистемное расстройство, возникающее в раннем детском возрасте, при котором наблюдаются признаки врожденного дискератоза наряду с гипоплазией мозжечка. У пациентов наблюдается классическая диагностическая триада врожденного дискератоза, в дополнение к отставанию в развитии, ЗВУР и недостаточности костного мозга.
Синдром Хойераала-Хрейдарссона генетически гетерогенен и вызван Х-сцепленными рецессивными мутациями в гене DKC1. Некоторые пациенты могут также страдать от тяжелого иммунодефицита. Синдром Ревеса проявляется в раннем детстве и имеет много общего с врожденным дискератозом. Диагноз подтверждается при наличии двусторонней экссудативной ретинопатии. У пациентов могут также наблюдаться очаги в/черепной кальцификации, ЗВУР, отставание в развитии и недостаточность костного мозга. При синдроме Ревеса мутирует ген TINF2, что относит это заболевание в основном к АуД-состояниям, но были описаны несколько случаев без идентифицированной мутации. У людей с этими тяжелыми формами врожденного дискератоза длина теломер даже короче, чем у людей с классическим вариантом врожденного дискератоза.
Синдром Коутса плюс вызван сложными гетерозиготными мутациями в гене СТС1 и имеет общие черты с врожденным дискератозом, включая редкие и седеющие волосы, дистрофические ногти и анемию.
Теломеры очень короткие. Синдром Коутса плюс характеризуется телеангиэктазией сетчатки и экссудатами в сетчатке, в/черепной кальцификацией, лейкодистрофией, кистами ГМ, остеопенией, ЖКК и портальной гипертензией, вызванной развитием сосудистых эктазий в желудке, тонком кишечнике и печени.
6. Осложнения. Примерно у 10-15% пациентов с врожденным дискератозом, как правило, на 3-м и 4-м десятилетиях жизни, развиваются ЗНО. Пациенты с врожденным дискератозом предрасположены к миелодиспластическому синдрому и острому миелоидному лейкозу, а также к солидным опухолям. 40% случаев рака у таких больных — это плоскоклеточный рак органов головы и шеи (язык, рот, глотка). Часто встречаются сквамозно-клеточная карцинома кожи и ЖКТ (пищевод, желудок, толстая кишка), а также аноректальная аденокарцинома.
Риск развития миелодиспластического синдрома увеличивается с возрастом и в 2362 раза выше, чем в общей популяции. Актуарный риск развития клональных и злокачественных миелоидных заболеваний составляет 25% к 18 годам. Др. опасные для жизни осложнения включают фиброз легких, фиброз печени и сильное ЖКК.
7. Лечение. Андрогены могут индуцировать улучшение функции костного мозга у 70% пациентов, а у некоторых пациентов в результате такого лечения трехлинейные показатели крови могут прийти в норму на несколько лет. Пациенты с врожденным дискератозом становятся невосприимчивыми к андрогенам по мере прогрессирования апластической анемии. Они также имеют тенденцию к большей чувствительности к побочным эффектам андрогенов, чем пациенты с АФ, поэтому важно начинать терапию с более низких доз и необходимо проводить частый мониторинг побочных эффектов.
Когда ответ не терапию максимален, доза андрогена м.б. медленно и постепенно снижена до минимальной дозы, необходимой для поддержания желаемого и безопасного количества клеток крови, но терапия не м.б. остановлена полностью.
Информации о применении иммуносупрессивной терапии для пациентов с врожденным дискератозом опубликовано мало, но есть единичные сообщения о нескольких пациентах, которым был неверно поставлен диагноз приобретенной апластической анемии и которые проходили иммуносупрессивную терапию без ответа. Цитокиновая терапия с гранулоцитарно-макрофагальным колониестимулирующим фактором или только с гранулоцитарным колониестимулирующим фактором или в сочетании с эритропоэтином, по-видимому, потенциально эффективна, по крайней мере в краткосрочной перспективе, но информации пока недостаточно.
Использование цитокинов должно быть сбалансировано с потенциальным стимулирующим действием этих препаратов на еще не обнаруженные клетки миелодиспластического синдрома или острого миелоидного лейкоза.
Аллогенная трансплантация гемопоэтических стволовых клеток является единственным методом радикального лечения при тяжелой недостаточности костного мозга, миелодиспластическом синдроме и остром миелоидном лейкозе, долгосрочная выживаемость, даже у родственных HLA-совместимых доноров гемопоэтических стволовых клеток, является низкой и составляет ок. 50%. Заболеваемость и смертность являются результатом осложнений, связанных с трансплантацией, таких как отторжение трансплантата, РТПХ, сепсис или веноокклюзионная болезнь, а также результатом осложнений, связанных с врожденным дискератозом, таких как фиброз легких и ЖКК, связанных с сосудистыми аномалиями.
Смертность, связанная с трансплантацией гемопоэтических стволовых клеток, выше, чем при др. синдромах врожденной недостаточности костного мозга, и, вероятно, вызвана высоким количеством легочно-сосудистых осложнений, наблюдаемых у пациентов с врожденным дискератозом, которые ассоциированы с лежащим в основе дефектом поддержания длины теломер.
Хотя мутации генов, лежащие в основе большинства случаев врожденного дискератоза известны, генная терапия данного заболевания пока находится в отдаленной перспективе.
8. Прогноз. Врожденный дискератоз в значительной степени является генетически гетерогенным заболеванием, кроме того, имеются некоторые данные о корреляциях генотипа и фенотипа. У пациентов с мутациями в определенных группах генов (напр., TERC, TERT) может развиться тяжелая апластическая анемия или фиброз печени и легких, но эти осложнения могут проявиться в более поздний период жизни и могут не сопровождаться мультисистемным поражением. У пациентов с мутациями в др. группах генов (напр., DKC1, TINF2, PARN, ACD, RTEL1), по-видимому, наблюдается больше физических аномалий и более высокая частота заболеваемости с более ранним началом апластической анемии и рака. Средний возраст смерти пациентов с врожденным дискератозом, диагностированным в детском возрасте, составляет ~30 лет.
Основными причинами смерти являются недостаточность костного мозга, осложнения трансплантации гемопоэтических стволовых клеток, онкологические заболевания, несовместимые с жизнью нарушения в легких и ЖКК.
г) Врожденная амегакариоцитарная тромбоцитопения:
1. Этиология и эпидемиология. Врожденная амегакариоцитарная тромбоцитопения встречается реже, чем анемия Фанкони, синдром Швахмана-Даймонда и врожденный дискератоз. Она передается АуР-путем. Врожденная амегакариоцитарная тромбоцитопения обычно проявляется в младенчестве в виде изолированной тромбоцитопении, вызванной снижением количества или отсутствием мегакариоцитов костного мозга с первоначальным сохранением гранулопоэтических и эритроидных линий. Панцитопения вследствие апластической анемии часто возникает в первые несколько лет жизни. Сообщалось о развитии миелодиспластического синдрома и острого миелоидного лейкоза у пациентов с врожденной амегакариоцитарной тромбоцитопенией и персистирующей апластической анемией.
Патология при врожденной амегакариоцитарной тромбоцитопения напрямую связана с мутациями в гене MPL, т.е. в гене рецептора тромбопоэтина. Тромбопоэтин — это фактор роста, который способствует выживанию гемопоэтических стволовых клеток и стимулирует пролиферацию и созревание мегакариоцитов. Гетерозиготы мутантного гена имеют нормальную гематологию, тогда как пораженные особи имеют мутации в обоих аллелях. Основываясь на генотип-фенотипических корреляциях, можно предсказать течение и сделать прогноз заболевания. Нонсенс-мутации вызывают полную потерю функции рецептора тромбопоэтина, что приводит к устойчиво низкому количеству тромбоцитов в раннем младенчестве из-за отсутствия мегакариоцитов и быстрому прогрессированию до панцитопении и апластической анемии (врожденная амегакариоцитарная тромбоцитопения типа I).
Выживаемость пораженных стволовых клеток при нонсенс-мутациях гена MPL объясняет прогрессирование врожденной амегакариоцитарной тромбоцитопении в апластическую анемию, поскольку тромбопоэтин также оказывает антиапоптотический эффект на гемопоэтические стволовые клетки, способствуя выживаемости клеток. Миссенс-мутации MPL связаны с более мягким течением заболевания, более поздним проявлением, частичным и преходящим увеличением количества тромбоцитов в течение первого года жизни после выявления заболевания и отсроченным началом панцитопении, если таковая имеется, указывающей на остаточную рецепторную функцию (врожденная амегакариоцитарная тромбоцитопения тип II). Биологически активный тромбопоэтин плазмы крови стабильно повышен у всех пациентов с врожденной амегакариоцитарной тромбоцитопенией.
2. Клинические проявления. У пациентов с врожденной амегакариоцитарной тромбоцитопенией наблюдается петехиальная сыпь, кровоподтеки или кровотечения. Начало проявления симптомов может зависеть от тяжести мутаций и колеблется от рождения до первого года жизни. Физические параметры и результаты диагностической визуализации большинства пациентов с врожденной амегакариоцитарной тромбоцитопенией находятся в норме. Ок. 10-20% опубликованных фенотипических случаев врожденной амегакариоцитарной тромбоцитопенией связаны с физическими аномалиями.
Наиболее распространенными аномалиями являются неврологические и кардиологические. Часто встречаются случаи, связанные с атрофией мозжечка и ГМ, кроме того, характерной особенностью заболевания является отставание в развитии. ВПС включают ДМПП и ДМЖП, ОАП, тетраду Фалло и коарктацию аорты. Некоторые из нарушений встречаются совместно.
Др. аномалии включают аномалии бедер или ступней, пороки развития почек, аномалии глаз и расщелину или арковидное небо. У некоторых пациентов наблюдается микроцефалия и лицевые патологии.
3. Результаты лабораторных исследований. Основным признаком врожденной амегакариоцитарной тромбоцитопенией по данным лабораторных исследований является тромбоцитопения, наблюдаемая первоначально при нормальных уровнях Hb и лейкоцитов. Количество тромбоцитов в периферической крови снижено или тромбоциты полностью отсутствуют. Как и при др. синдромах врожденной недостаточности костного мозга, эритроциты м.б. макроцитарными. Уровень HbF м.б. повышен, а также может наблюдаться повышенная экспрессия АГн i. Исходные аспираты и биопсия костного мозга показывают нормальную клеточность с выраженным снижением количества мегакариоцитов или их отсутствием. У пациентов с развивающейся апластической анемией, клеточность костного мозга снижается, сопровождаясь жировым перерождением; эритропоэтические и гранулопоэтические линии также симметрично уменьшаются.
4. Диагностика. Аспирация и биопсия костного мозга показаны, если тромбоцитопения сохраняется и после неонатального периода или ассоциирована с адекватным ответом на гемотрансфузию тромбоцитов и не выявлено очевидного фактора, способствующего преципитации, такого как инфекции или иммунологические реакции. Дефицит мегакариоцитов в таких случаях указывает на диагноз, а мутационный анализ его подтверждает. Если врожденная амегакариоцитарная тромбоцитопения возникает при рождении или вскоре после него, то это заболевание следует дифференцировать от др. причин наследственной и приобретенной неонатальной тромбоцитопении. Тромбоцитопения с отсутствием лучевых костей (TAR-синдром) отличается от врожденной амегакариоцитарной тромбоцитопенией тем, что при тромбоцитопении с отсутствием лучевых костей отсутствуют лучевые кости.
Отличие от врожденного дискератоза может проявляться отсутствием кожно-слизистых, неврологических и иммунологических аномалий, характерных для форм врожденного дискератоза с ранним проявлением. Значения длины теломер ниже 1-го процентиля, наблюдаемых у сопоставимых по возрасту здоровых контролей, характерны для врожденного дискератоза, но не для врожденной амегакариоцитарной тромбоцитопенией. И наконец, лимфоциты крови при врожденной амегакариоцитарной тромбоцитопенией не демонстрируют повышенного количества хромосомных разрывов при воздействии диэпоксибутана, что отличает это заболевание от анемии Фанкони.
5. Осложнения. У некоторых пациентов появляются клональные цитогенетические аномалии костного мозга, такие как моносомия 7 и трисомия 8 хромосом. Врожденная амегакариоцитарная тромбоцитопения может прогрессировать в миелодиспластический синдром и острый лейкоз, но истинный риск не м.б. определен из-за редкости заболевания и недостаточности опубликованных данных.
6. Терапия и прогноз. Смертность от тромбоцитопенических кровотечений, осложнений апластической анемии или лейкозной трансформации у пациентов с нонсенс-мутациями гена MPL близка к 100%, если функция костного мозга не улучшается. Пациенты с миссенс-мутациями имеют более мягкое течение, но у них также могут наблюдаться серьезные осложнения. Трансплантация гемопоэтических стволовых клеток — это единственный возможный метод радикального лечения. Большинство пациентов с врожденной амегакариоцитарной тромбоцитопенией, которым проводится трансплантация гемопоэтических стволовых клеток, излечиваются, особенно если процедура проводится с HLA-совместимыми родственными донорами. Гемотрансфузию тромбоцитов перед трансплантацией следует проводить дискретно.
Количество тромбоцитов не всегда должно быть единственным показанием для лечения, но такие симптомы, как клиническое проявление кровотечения, являются достаточным сигналом. Для минимизации сенсибилизации предпочтительны лейкоредуцированные тромбоциты, взятые от одного донора. Продукты крови для всех кандидатов на трансплантацию гемопоэтических стволовых клеток должны быть облучены и проверены на ЦМВ.
Кортикостероиды не являются эффективным средством лечения тромбоцитопении. Нет данных, подтверждающих успешное применение андрогенов для временного улучшения апластической анемии. Тромбомиметики, такие как элтромбопаг или ромиплостим, могут подойти некоторым пациентам (врожденная амегакариоцитарная тромбоцитопения типа II), поэтому необходимо их дальнейшее изучение. И все же, из-за того, что вышеперечисленные агенты стимулируют фиброз, а также из-за риска развития миелодиспластического синдрома и лейкоза при врожденной амегакариоцитарной тромбоцитопении, трансплантация гемопоэтических стволовых клеток остается предпочтительным методом лечения пациентов с тяжелой цитопенией.
д) Другие виды врожденной апластической анемии. С появлением методов скрининга всего генома было идентифицировано значительное число генов, ассоциированных с недостаточностью костного мозга при панцитопении (см. табл. 1). Специфические генетические расстройства могут варьироваться по фенотипу, но часто характеризуются физическими пороками развития, семейным анамнезом, ранним возрастом начала заболевания, панцитопенией и риском развития миелодиспластического синдрома и острого миелоидного лейкоза. Наследственные синдромы панцитопении и семейные синдромы миелодиспластического синдрома и острого миелоидного лейкоза очень похожи между собой.
1. Ретикулярный дисгенез. Ретикулярный дисгенез — это вариант тяжелого комбинированного иммунодефицита с врожденным агранулоцитозом. Также могут наблюдаться анемия и тромбоцитопения. Ретикулярный дисгенез вызван гомозиготными или сложными гетерозиготными мутациями в гене митохондриальной аденилаткиназы-2 (АК2) на хромосоме 1р35. Возможно участие др. генов и др. пути наследования, которые м.б. выявлены в будущем. При ретикулярном дисгенезе отсутствует клеточный и гуморальный иммунитет, также наблюдаются выраженная лимфопения и нейтропения. Степени анемии и тромбоцитопении различны, иногда развивается тяжелая апластическая анемия. Образцы костного мозга гипоцеллюлярные, с сильно сниженным количеством миелоидных и лимфоидных элементов. Единственный метод радикального лечения — это трансплантация гемопоэтических стволовых клеток.
2. Хрящево-волосяная гипоплазия. Хрящево-волосяная гипоплазия — это АуР-синдром, который встречается, в основном, среди популяций финнов или амишей. Он характеризуется метафизарным дизостозом, карликовостью с короткими конечностями и тонкими, редкими волосами. Др. признаки со стороны скелетной системы: сколиоз, лордоз, деформация ГК и варусная деформация нижних конечностей. Также встречаются аномалии ЖКТ. Хрящево-волосяная гипоплазия вызывается мутациями в гене RMRP. Макроцитарная анемия наблюдается у большинства пациентов и иногда бывает тяжелой и персистирующей. Нейтропения, лимфопения и предрасположенность к лимфоме и др. им опухолям также являются характерными особенностями данного заболевания. Радикальной терапией является трансплантация гемопоэтических стволовых клеток.
е) Другие наследственные синдромы с периодической выраженной недостаточностью костного мозга:
1. Синдром Дауна. Синдром Дауна (трисомия по хромосоме 21) особым образом ассоциирована с аберрантными гематологическими показателями. Больные имеют склонность к острым лимфобластным и миелобластным лейкозам, особенно к острым мегакариобластным лейкозам. Сообщалось о редких пациентах с синдромом Дауна и панцитопенией, вызванной апластической анемией.
2. Синдром Дубовица. Синдром Дубовица — это АуР-заболевание, характеризующееся лицевыми патологиями, детской экземой, небольшим ростом и легкой микроцефалией. Лицо небольшое, с гипопластической надбровной дугой, переносицей, расположенной на уровне лба, короткими щелями век, птозом и микрогнатией. У этих пациентов есть предрасположенность к онкологическим заболеваниям. У 10% пациентов наблюдаются нарушения кроветворения, включая умеренную панцитопению, гипопластическую анемию, гипоплазию костного мозга и резко выраженную апластическую анемию. Синдром Дубовица связан с мутациями в генах NSUN2 (РНК-метилтрансферазе) и LIG4 (ядерной ДНК-лигазе).
3. Синдром Секеля. Синдром Секеля (SCKL), иногда называемый «птицеголовой карликовостью», представляет собой АуР-нарушение развития, характеризующееся выраженным отставанием в росте и умственной отсталостью, микроцефалией, гипопластическим лицом с выступающим носом и низко посаженными и/или деформированными ушами. У 25% пациентов наблюдается апластическая анемия или ЗНО. Существует широкая генетическая гетерогенность, включающая по меньшей мере 8 классифицируемых типов: SCKL1 с мутацией в гене ATR; SCKL2 с мутацией в гене RBBP8; SCKL3 с мутацией в гене 14q21-q22; SCKL4 с мутацией в гене CENPJ; SCKL5 с мутацией в гене СЕР 152; SCKL6 с мутацией в гене СЕР63; SCKL7 с мутацией в гене NIN и SCKL8 с мутацией в гене ATRIP.
4. Иммунокостная дисплазия Шимке. Иммунокостная дисплазия Шимке — это АуР-заболевание, вызванное мутациями в белке ремоделирования хроматина SMARCAL1. У пациентов наблюдается спондилоэпифизарная дисплазия с ненормально увеличенным поясничным лордозом и выступающим животом. Наблюдаются пигментные изменения кожи и аномально обесцвеченные и сформированные зубы. Нарушение функции почек м.б. критичным и сопровождаться протеинурией и нефротическим синдромом. У 50% пациентов наблюдается гипотиреоз; у 50% — ишемия ГМ; у 10% — недостаточность костного мозга с нейтропенией, тромбоцитопенией и анемией; и ~5% предрасположены к не-ходжкинской лимфоме. Практически у всех пациентов наблюдается лимфопения и нарушение клеточного иммунитета. В двух опубликованных описаниях клинических случаев 2 пациента успешно перенесли трансплантацию костного мозга.
5. Синдром Нунан. Синдром Нунан — это нарушение развития, характеризующееся особыми чертами лица (гипертелоризм, птоз, короткая шея, низкорасположенные ушные раковины), низким ростом, врожденным пороком сердца и множественными скелетными и гематологическими аномалиями. Это, в первую очередь, АуД-расстройство, включающее несколько генетических типов (см. табл. 1). Гетерозиготные мутации в гене PTPN11 вызывают ок. 50% случаев синдрома Нунан. Др. случаи вызваны мутациями в др. генах, ассоциированных с сигнальным путем RAS, таких как KRAS, SOS1 и NRAS. Также были выявлены АуР-формы, вызываемые мутациями в генах SHOC2 или CBL. Помимо ассоциации с ювенильным миеломоноцитарным лейкозом, у пациентов с синдромом Нунан может развиться амегакариоцитарная тромбоцитопения, а также панцитопения с гипоцеллюлярным костным мозгом.
ж) Неклассифированные синдромы врожденной недостаточности костного мозга. Неклассифицированные синдромы врожденной недостаточности костного мозга — это гетерогенные нарушения, которые могут представлять собой атипичное проявление либо известных заболеваний, либо новых синдромов. Данное определение было доказано с помощью объективных методов, таких как комплексные панели всех известных генов синдромов врожденной недостаточности костного мозга независимо от специфических гематологических (напр., изолированная нейтропения или панцитопения) или негематологических проявлений или секвенирование всего экзома / генома.
Эти нарушения не вписываются в классическую генетическую патологию недостаточности костного мозга, поскольку все признаки заболевания м.б. не выражены при проявлении заболевания. Все они характеризуются разл. цитопениями, вызванными непродуктивным костным мозгом с соматическими проявлениями или без них. По сравнению с классическими вариантами (проявление в возрасте ~1 мес), неклассифицированные нарушения проявляются позже (в возрасте 9 мес) и проявляются однолинейной или многолинейной цитопенией, апластической анемией, миелодисплазией или ЗНО с разл. пороками развития. В табл. 4 перечислены критерии для постановки диагноза, которые включают признаки хронической недостаточности костного мозга, а также факторы, указывающие на высокую вероятность наследственного заболевания (напр., семейный анамнез, врожденные аномалии, ранний возраст при проявлении заболевания).
Если заболевание проявляется позже и без физических пороков развития, не м.б. исключен приобретенный характер заболевания. Детальное генетическое тестирование на наличие известных генов синдрома врожденной недостаточности костного мозга или тестирование с помощью секвенирования всего экзома/генома может выявить наследственную этиологию. Кроме того, было показано, что при некоторых синдромах во время последующего наблюдения проявляются типичные физические особенности.
Определение фактической генетической причины помогает классифицировать пациентов в соответствии с болезнью и способствует правильному консультированию и надлежащему мед. обслуживанию. Во многих случаях к выполнению плана лечения следует приступать немедленно. Лечение таких пациентов должно осуществляться в соответствии с типом осложнений, которые наблюдаются у пациента при проявлении заболевания, а также в соответствии с научным опытом, опубликованным по неклассифицированным случаям.