а) Дефицит пируваткиназы. Врожденная гемолитическая анемия возникает у гомозиготных/сложных гетерозиготных пациентов по АуР-генам, которые вызывают либо выраженное снижение уровня пируваткиназы в эритроцитах, либо выработку аномального фермента со сниженной активностью, приводящего к нарушению превращения фосфоенолпирувата в пируват.
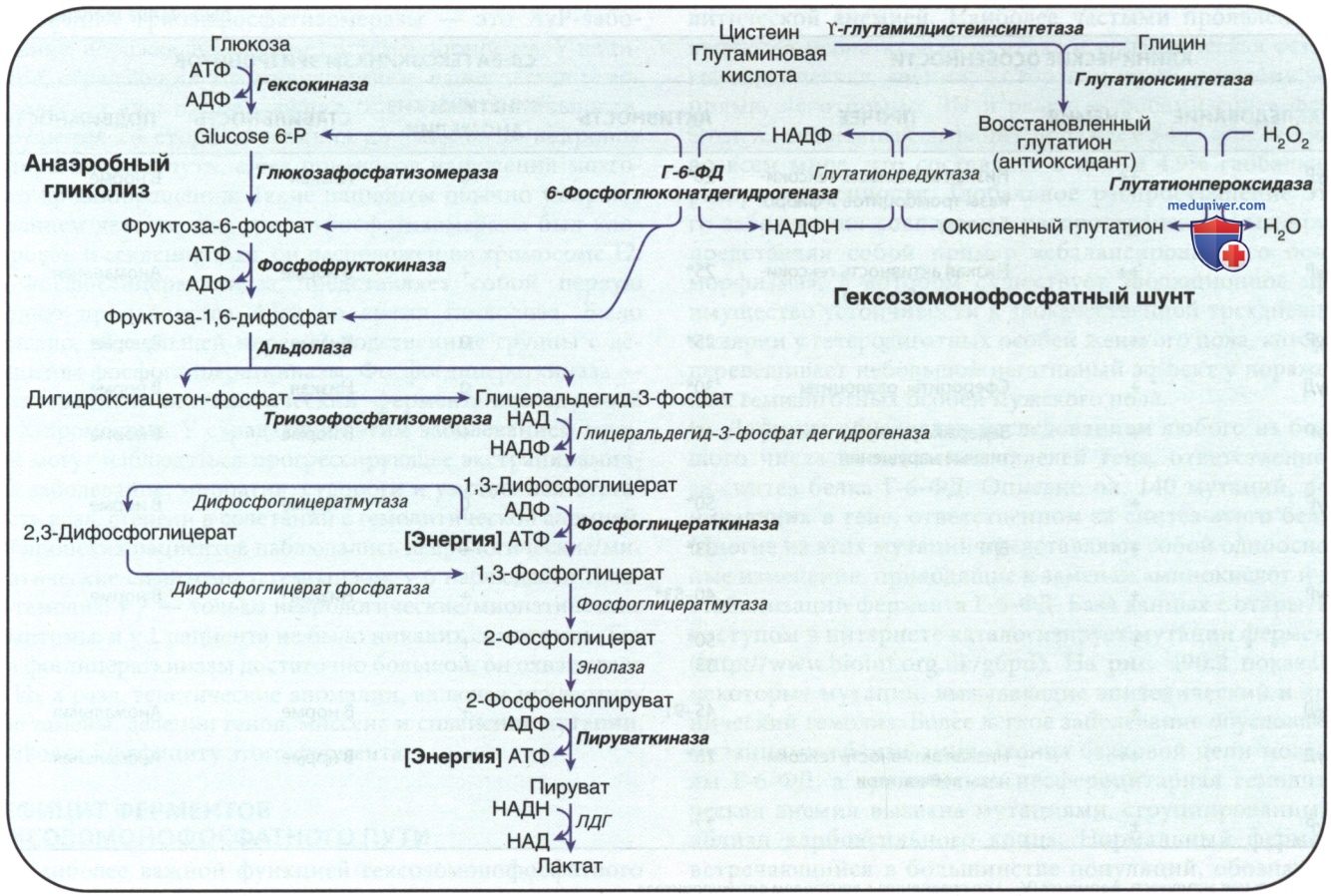
Образование АТФ в эритроцитах на этой стадии нарушается, наблюдаются низкие уровни АТФ, пирувата и окисленной формы НАД+ (рис. 1). Концентрация 2,3-дифосфоглицерата повышается; этот изомер помогает выделять кислород из НЬ, но мешает ингибированию гексокиназы и ферментов гексозомонофосфатного шунта. Кроме того, происходит необъяснимое уменьшение общего количества адениновых (АТФ, АДФ и АМФ) и пиридиновых (НАД+ и восстановленная форма НАД) нуклеотидов, что еще больше нарушает гликолиз.
Рисунок 1. Метаболизм эритроцитов: гликолиз и гексозомонофосфатный путь. Дефицит ферментов, явно связанный с гемолизом, выделен жирным шрифтом. НАДФН — восстановленная форма никотинамидадениндинуклеотидфосфата
В результате снижения АТФ содержание калия и воды в эритроцитах не может поддерживаться на нормальном уровне; клетки становятся ригидными, а продолжительность их жизни значительно сокращается.
1. Этиология. Существует 2 гена пируваткиназы млекопитающих, но только ген PKLR (пируваткиназы печени и почек — прим. перевод.) экспрессируется в эритроцитах. Ген PKLR человека расположен на хромосоме 1q21. В этом структурном гене, кодирующем белок из 574 аминокислот и образующий функциональный тетрамер, зарегистрировано >180 мутаций. Эти мутации включают миссенс-мутации, мутации сайта сплайсинга и инсерционно-делеционные изменения, наблюдается некоторая корреляция типа, местоположения и аминокислотной замены с тяжестью заболевания.
Большинство пациентов, страдающих от этого нарушения, являются сложными гетерозиготами по 2 разл. дефектам гена пируваткиназы. Вариабельность клинической тяжести объясняется, вероятно, возможностью многочисленных комбинаций. Мутации 1456 С на Т и 1529 G на А являются наиболее распространенными мутациями среди белого населения.
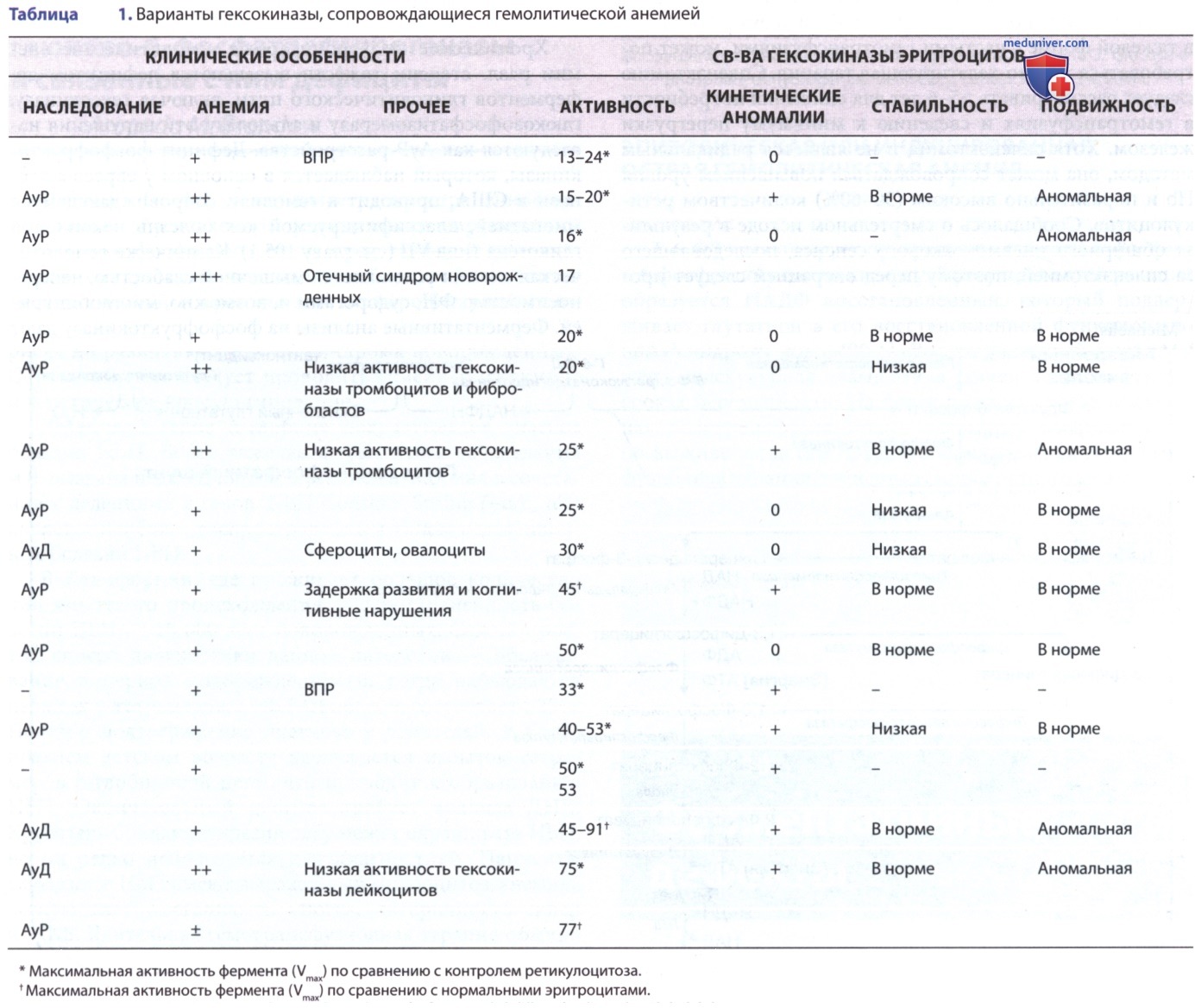
2. Клинические проявления и лабораторные данные. Клинические проявления дефицита пируваткиназы варьируют от тяжелой неонатальной гемолитической анемии до легкого, хорошо компенсированного гемолиза, впервые наблюдаемого в зрелом возрасте (табл. 1). Желтуха и анемия тяжелой степени могут возникнуть в неонатальном периоде, сообщалось о керниктерусе (ядерная желтуха). Гемолиз у детей старшего возраста и взрослых различается по степени тяжести, причем показатели Hb колеблются в пределах 8-12 г/дл, что сопровождается некоторой бледностью, желтухой и спленомегалией.
Количество ретикулоцитов часто чрезвычайно повышено, что отражает тяжелый длительный гемолиз. Пациенты с такими лабораторными показателями обычно не нуждаются в гемотрансфузии. Тяжелая форма этого нарушения имеет относительно высокую заболеваемость у представителей амишей (амиши — последователи одной из протестантских сект в США) Среднего Запада США. Дефицит пируваткиназы, возможно, обеспечивает защиту организма от злокачественной трехдневной малярии.
Полихроматофилия и умеренный макроцитоз отражают повышенное количество ретикулоцитов. Сфероциты встречаются редко, но можно обнаружить несколько шиповидных пикноцитов. Диагноз ставится на основании выраженного снижения активности пируваткиназы в эритроцитах/увеличении константы диссоциации Михаэлиса-Ментена (Michaelis, Menten) (Km) для его субстрата — фосфоенолпирувата (вариант с высоким значением Km).
Активность др. ферментов эритроцитов нормальная/повышенная, что отражает ретикулоцитоз. Никаких отклонений от нормы показателей Hb не отмечено. Пируваткиназа лейкоцитов имеет нормальную активность, поэтому последние должны быть строго исключены из гемолизатов эритроцитов, используемых для измерения активности фермента. Гетерозиготные носители обычно имеют умеренно сниженный уровень активности пируваткиназы.
3. Лечение. При гипербилирубинемии у новорожденных м.б. показаны фототерапия и заместительные гемотрансфузии. При тяжелой анемии/апластических кризах необходима гемотрансфузия эритроцитарной массы. Если анемия протекает в тяжелой форме с частыми гемотрансфузиями, может потребоваться железо-хелатирующая терапия. Спленэктомию следует рассматривать >5-6 лет для снижения потребности в гемотрансфузиях и сведению к минимуму перегрузки железом. Хотя спленэктомия и не является радикальным методом, она может сопровождаться повышением уровня Hb и поразительно высоким (30-60%) количеством ретикулоцитов.
Сообщалось о смертельном исходе в результате обширного пневмококкового сепсиса, последовавшего за спленэктомией; поэтому перед операцией следует проводить вакцинацию против инкапсулированных микроорганизмов, а после в профилактических целях назначать пенициллин. Спленэктомия также обусловливает развитие тромбоза и легочной гипертензии. У любого пациента с врожденной гемолитической анемией и рецидивирующими болями в животе следует подозревать наличие камней в желчном пузыре. В настоящее время не существует радикальной терапии; фармакологический активатор пируваткиназы изучается в клинических испытаниях на начальном этапе.
Данные об естественной динамике заболевания ограничены и в настоящее время изучаются с помощью международного реестра.
б) Дефицит других гликолитических ферментов. Хронические несфероцитарные гемолитические анемии разл. степени тяжести вызываются дефицитом др. ферментов гликолитического пути, включая гексокиназу, глюкозофосфатизомеразу и альдолазу, эти нарушения наследуются как АуР-расстройства. Дефицит фосфофруктокиназы, который наблюдается в основном у евреев ашкенази в США, приводит к гемолизу, сопровождающемуся миопатией, классифицируемой как болезнь накопления гликогена типа VII.
Клинически гемолитическая анемия осложняется мышечной слабостью, непереносимостью ФН, судорогами и, возможно, миоглобинурией. Ферментативные анализы на фосфофруктокиназу дают низкие значения содержания ее в эритроцитах и мышцах.
Дефицит триозофосфатизомеразы — это АуР-забо-левание, поражающее многие системы организма. У пациентов, страдающих этим нарушением, наблюдается гемолитическая анемия, нарушения сердечной деятельности, нарушения со стороны нижних двигательных нейронов и пирамидного пути, с/без признаков нарушения мозгового кровообращения. Такие пациенты обычно умирают в раннем детстве. Ген триозофосфатизомеразы был клонирован и секвенирован, он расположен на хромосоме 12.
Фосфоглицераткиназа представляет собой первую стадию производства АТФ во время гликолиза. Было описано, по меньшей мере, 23 родственные группы с дефицитом фосфоглицераткиназы. Фосфоглицераткиназа — единственный гликолитический фермент, наследуемый по Х-хромосоме. У страдающих этим заболеванием мужчин могут наблюдаться прогрессирующее экстрапирамидное заболевание, миопатия, судороги и умственная отсталость разл. степени в сочетании с гемолитической анемией.
У 9 японских пациентов наблюдались неврологические/миопатические симптомы с гемолизом; у 6 наблюдался только гемолиз; у 7 — только неврологические/миопатические симптомы; и у 1 пациента не было никаких симптомов.
Ген фосфоглицераткиназы достаточно большой, он охватывает 23 kb, а разл. генетические аномалии, включая нуклеотидные замены, делеции генов, миссенс и сплайсинг мутации, приводят к дефициту этого фермента.
1. Дефицит ферментов гексозомонофосфатного пути. Наиболее важной функцией гексозомонофосфатного пути является поддержание глутатиона в его восстановленной форме в качестве защиты от окисления эритроцитов (см. рис. 1). Примерно 10% глюкозы, поглощаемой эритроцитами, проходит через этот путь для образования восстановленной формы НАДФ (НАДФН), необходимой для превращения окисленного глутатиона в восстановленную его форму. Поддержание восстановленной формы глутатиона необходимо для физиол. инактивации окислительных соединений, таких как перекись водорода, которые образуются в эритроцитах.
Если количество глутатиона/любого др. соединения/фермента, необходимого для поддержания его в восстановленной форме, сокращается, SH-группы мембран эритроцитов окисляются, Hb денатурируется и может осаждаться в виде включений в эритроцитах, называемых тельцами Гейнца (Heinz). После образования телец Гейнца начинается острый гемолитический процесс, который возникает в результате повреждения мембран эритроцитов выпавшим в осадок Hb, окислителем и селезенкой. Затем поврежденные эритроциты быстро выводятся из кровообращения.
в) Дефицит глюкозо-6-фосфатдегидрогеназы и связанные с ним дефициты. Дефицит Г-6-ФД — наиболее распространенное заболевание, обусловленное ферментными нарушениями гексозомонофосфатного пути, представлен 2 клиническими синдромами: эпизодической острой гемолитической анемией и хронической несфероцитарной гемолитической анемией. Наиболее частыми проявлениями являются неонатальная желтуха и эпизодическая острая гемолитическая анемия, которая индуцируется инфекциями, некоторыми ЛП и редко — бобами сорта фава.
Этот Х-сцепленный дефицит поражает >400 млн человек во всем мире, что составляет в целом 4,9% глобальной распространенности.
Глобальное распространение этого заболевания совпадает с распространением малярии, представляя собой пример «сбалансированного полиморфизма», в котором существует эволюционное преимущество устойчивости к злокачественной трехдневной малярии у гетерозиготных особей женского пола, которое перевешивает небольшой негативный эффект у пораженных гемизиготных особей мужского пола.
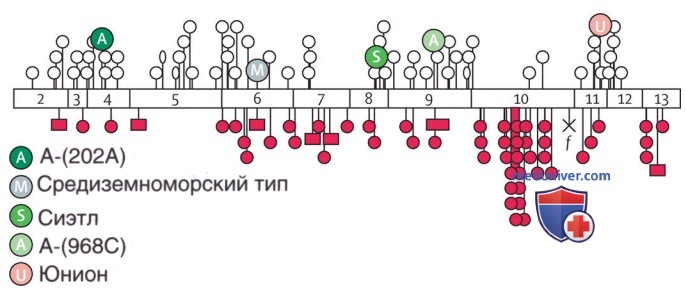
Дефицит обусловлен наследованием любого из большого числа аномальных аллелей гена, ответственного за синтез белка Г-6-ФД. Описано ок. 140 мутаций, возникающих в гене, ответственном за синтез этого белка. Многие из этих мутаций представляют собой одноосновные изменения, приводящие к заменам аминокислот и дестабилизации фермента Г-6-ФД. База данных с открытым доступом в интернете каталогизирует мутации фермента. На рис. 2 показаны некоторые мутации, вызывающие эпизодический и хронический гемолиз.
Рисунок 2. Наиболее распространенные мутации вдоль кодирующей последовательности гена глюкозо-6-фосфатдегидрогеназы. Экзоны показаны не закрашенными пронумерованными прямоугольниками. Не закрашенные круги — мутации, вызывающие варианты класса II и III. Закрашенные круги представляют собой спорадические мутации, приводящие к тяжелым вариантам (класс I). Не закрашенные эллипсы — мутации, вызывающие варианты класса IV. X — нонсенс-мутация; f — мутация сайта сплайсинга; закрашенные квадраты — небольшие делеции. 202А и 968С — 2 сайта замещения оснований в глюкозо-6-фосфатдегидрогеназе-А
Более легкое заболевание обусловлено мутациями вблизи амино-конца белковой цепи молекулы Г-6-ФД, а хроническая несфероцитарная гемолитическая анемия вызвана мутациями, сгруппированными вблизи карбоксильного конца.
Нормальный фермент, встречающийся в большинстве популяций, обозначается как Г-6-ФД В+. Нормальный вариант, обозначенный Г-6-ФД А+, распространен среди американцев африканского происхождения.
1. Эпизодическая или индуцированная острая гемолитическая анемия:
- Этиология. Г-6-ФД катализирует превращение глюкозы 6-фосфата в 6-фосфоглюконовую кислоту. В результате этой реакции образуется НАДФ восстановленный, который поддерживает глутатион в его восстановленной функциональной форме (см. рис. 1). Восстановленный глутатион обеспечивает защиту организма от угроз окислительных реакций со стороны некоторых ЛП и инфекций, которые в противном случае вызвали бы осаждение Hb (тельца Гейнца)/повреждение мембран эритроцитов.
Синтез Г-6-ФД в эритроцитах определяется геном на Х-хромосоме. Т.о., гетерозиготные пациенты женского пола обладают промежуточной ферментативной активностью и имеют две популяции эритроцитов: одну нормальную, а другую — с дефицитом активности Г-6-ФД. Поскольку у гетерозиготных особей женского пола меньше клеток, предрасположенных к заболеванию, у большинства из них не наблюдается клинически выраженного гемолиза после воздействия оксидативных ЛП.
В редких случаях у гетерозиготных женщин большая часть эритроцитов является Г-6-ФД-дефицитными, вследствие случайной и иногда слишком обширной инактивации нормальной Х-хромосомы (гипотеза Лайона-Бейтлера (Lyon, Beutler).
Поэтому заболевания, обусловленные патологией этого фермента, чаще встречаются у мужчин, чем у женщин. Приблизительно 13% мужчин-американцев африканского происхождения имеют мутантный фермент (Г-6-ФД А-), что приводит к дефициту активности Г-6-ФД в эритроцитах (5-15% от нормы). Итальянцы, греки и др. средиземноморские, ближневосточные, африканские и азиатские этнические группы также имеют высокую частоту заболеваемости, в пределах 5-40%, вариантом, обозначаемым Г-6-ФД В- (Г-6-ФД Средиземноморский тип). При этих вариантах активность Г-6-ФД у гомозиготных особей женского пола/гемизиготных особей мужского пола <5% от нормы.
Данный дефект имеет менее тяжелую форму у американцев африканского происхождения в сравнении с американцами европейского происхождения. Третий тип мутантного фермента со значительно сниженной активностью (Г-6-ФД Кантон) встречается у ~5% китайского населения.
- Клинические проявления. У большинства людей с дефицитом Г-6-ФД заболевание протекает бессимптомно, без клинических проявлений, если только такие проявления не вызваны инфекцией, ЛП и употреблением в пищу бобов сорта фава. Как правило, гемолиз возникает через ~24-48 ч после того, как пациент употребил в-во с оксидативными св-вами. В тяжелых случаях возникают гемоглобинурия и желтуха, а концентрация Hb может резко упасть. ЛП, вызывающие гемолиз у этих пациентов, включают ацетилсалициловую кислоту («Аспирин»), сульфаниламиды, расбуриказу и противомалярийные ЛП, такие как примахин (табл. 2).
Степень гемолиза зависит от провоцирующего агента, количества проглоченного в-ва и степени тяжести ферментативного дефицита. У некоторых людей употребление в пищу бобов сорта фава также вызывает острый, тяжелый гемолитический синдром, известный как фавизм. Бобы фава содержат дивицин, изоурамил и конвицин, которые в конечном итоге приводят к образованию перекиси водорода и др. активных производных кислорода. Считается, что фавизм чаще бывает при В-варианте Г-6-ФД.
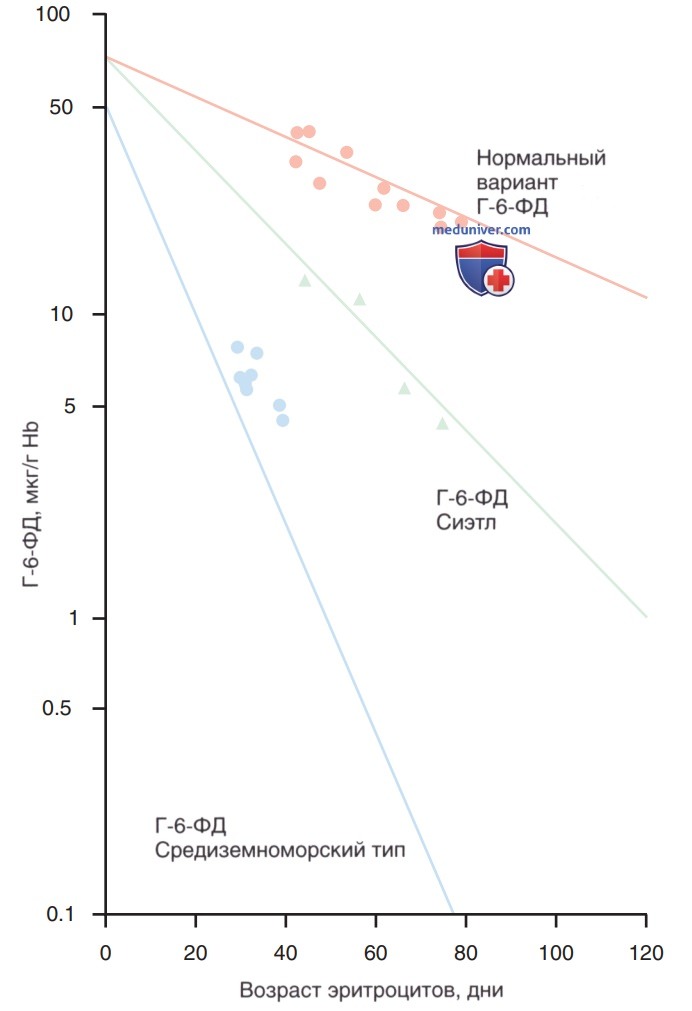
При варианте Г-6-ФД А стабильность димера свернутого белка нарушена, а дефект становится более выраженным по мере старения эритроцитов. Т.о., гемолиз уменьшается по мере разрушения старых эритроцитов, даже если введение ЛП продолжается. Это восстановление происходит за счет фермента, лабильного в зависимости от возраста, находящегося в избытке и являющегося стабильным в более молодых эритроцитах (рис. 3). Обусловленный этим ретикулоцитоз вызывает компенсированный гемолитический процесс, при котором уровень НЬ в крови м.б. лишь незначительно снижен, несмотря на продолжающееся воздействие провоцирующего агента.
Рисунок 3. Основной механизм развития дефицита глюкозо-6-фосфатдегидрогеназы в эритроцитах in vivo в условиях нестабильности мутантного фермента. При многих вариантах дефицита глюкозо-6-фосфатдегидрогеназы, напр., при двух вариантах, описанных здесь, это явление представляет собой ускорение процесса, обычно происходящего по мере старения эритроцитов во время циркуляции
Дефицит Г-6-ФД может вызывать гемолиз в неонатальном периоде. У недоношенных детей с Г-6-ФД А- наблюдались спонтанный гемолиз и гипербилирубинемия. У новорожденных с вариантами Г-6-ФД В- и Г-6-ФД Кантон может наблюдаться гипербилирубинемия и даже керниктерус. Новорожденные с сочетанным наследованием дефицита Г-6-ФД и мутацией промотора уридиндифосфат-глюкуронилтрансферазы, наблюдаемой при синдроме Жильбера (Gilbert), имеют более тяжелую форму неонатальной желтухи.
Если беременная женщина принимает оксидативные ЛП, они могут воздействовать на плод с дефицитом Г-6-ФД, и гемолитическая анемия и желтуха проявятся при рождении.
- Результаты лабораторных исследований. Начало острого гемолиза обычно приводит к резкому падению уровней Hb и Ht. Если эпизод тяжелый, то Hb-связывающие белки, такие как гаптоглобин, сатурируются, и свободный Hb может появиться в плазме и впоследствии в моче. Неокрашенные/суправитальные эритроцитарные препараты обнаруживают осажденный Hb/тельца Гейнца. При окраске мазка крови красителем Райта (Wright) включения эритроцитов не видны. Клетки, содержащие эти включения, видны только в первые 3-4 дня болезни, так как они быстро удаляются из крови.
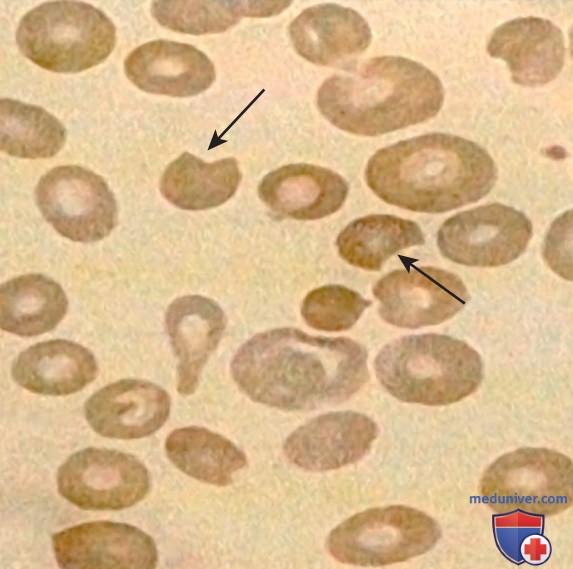
Кроме того, в мазке крови можно обнаружить эритроциты, кажущиеся надкусанными по периметру («надкусанные клетки»/дегмациты), и полихромазию (наличие синеватых, более крупных эритроцитов), свидетельствующих о ретикулоцитозе (рис. 4).
Рисунок 4. Морфологические изменения эритроцитов (анизопойкилоцитоз, «надкусанные» клетки) во время острого гемолиза у пациента с дефицитом глюкозо-6-фосфатдегидрогеназы. Стрелками показаны «надкусанные» клетки. Анизопойкилоцитоз — аномалия формы/раз-мера эритроцитов
- Диагностика. Постановка диагноза зависит от прямого/косвенного выявления сниженной активности Г-6-ФД в эритроцитах. При прямом измерении активность фермента у пациентов составляет <10% от нормы, при этом снижение активности фермента более выражено у американцев европейского происхождения и у азиатов, нежели у американцев африканского происхождения. Скрининговые тесты, отвечающие требованиям, основаны на обесцвечивании метиленового синего, снижении уровня метгемоглобина и флуоресценции восстановленного НАДФ.
Сразу после гемолитического эпизода преобладают ретикулоциты и молодые эритроциты. Эти молодые клетки обладают значительно более высокой ферментативной активностью, чем старые клетки при варианте А (у пациентов африканского происхождения). Поэтому тестирование откладывается на несколько недель до появления диагностически низкого уровня фермента. Диагноз можно заподозрить, если активность Г-6-ФД находится на нижней границе нормы при одновременно высоком ретикулоцитозе. Варианты дефицитов Г-6-ФД м.б. установлены посредством электрофоретического и молекулярного анализа. Дефицит фермента следует рассматривать у всех пациентов в неонатальном периоде при гипербилирубинемии и погранично низкой активности Г-6-ФД.
- Профилактика и лечение. Профилактика гемолиза является важнейшей терапевтической мерой. Мужчины, принадлежащие к этническим группам со значительной частотой заболеваемости дефицитом Г-6-ФД (напр., греки, южные итальянцы, евреи сефарды, филиппинцы, южные китайцы, американцы африканского происхождения, тайцы), должны быть, по возможности, обследованы на наличие дефекта до назначения определенных оксидативных ЛП. Обычные дозы ацетилсалициловой кислоты («Аспирина») и триметоприма/сульфаметоксазола не вызывают клинически значимого гемолиза при варианте А. Ацетилсалициловая кислота («Аспирин»), вводимый в дозах, используемых при острой ревматической лихорадке (60-100 мг/кг в сутки), может вызвать тяжелый гемолитический эпизод.
Младенцы с тяжелой неонатальной желтухой, относящиеся к вышеперечисленным этническим группам, нуждаются в тестировании на дефицит Г-6-ФД вследствие повышенного риска развития данного дефекта. Если возник тяжелый гемолиз, может потребоваться проведение гемотрансфузии, хотя выздоровление возможно только при прекращении приема оксидативного ЛП.
2. Хронические гемолитические анемии, связанные с дефицитом глюкозо-6-фосфатдегидрогеназы или родственных факторов. Хроническая несфероцитарная гемолитическая анемия вызвана тяжелым дефицитом Г-6-ФД, обусловленным разл. вариантами ферментов, особенно дефективными относительно количества, активности и стабильности. Дефекты генов, приводящие к хроническому гемолизу, локализуются преимущественно в области участка НАДФ-связывания вблизи карбоксильного конца белка (см. рис. 2). К таким дефектам относятся варианты Лома-Линда, Тома, Айова, Беверли-Хиллз, Нэшвилл, Риверсайд, Сантьяго-де-Куба и Андалус.
У людей с дефицитом фермента Г-6-ФД В иногда наблюдается хронический гемолиз, а гемолитический процесс может усугубляться после приема оксидативных ЛП. Спленэктомия мало эффективна при этих видах хронического гемолиза.
Др. ферментативные дефекты могут нарушать регенерацию восстановленного глутатиона в качестве оксидативного «зумпфа» (см. рис. 1). Сообщалось о легкой хронической несфероцитарной анемии в сочетании со снижением уровня восстановленного глутатиона в эритроцитах в результате дефицитов γ-глутамилцистеина/глутатион-синтетазы. Дефицит Г-6-ФД обусловливает главным образом медикаментозный гемолиз, а гемолиз с развитием гипербилирубинемии у новорожденных вызван дефицитом глутатион-пероксидазы.