Кистозные поражения печени м.б. первоначально выявлены в младенчестве и детстве. Фиброз печени также может возникать как проявление сопутствующего дефекта развития (табл. 1). Кистозная болезнь почек ассоциирована с кистозными поражениями печени и ЖВП и часто определяет клиническую картину и прогноз.
Практически все белки, кодируемые генами, мутировавшими при комбинированных кистозных заболеваниях печени и почек, частично локализуются в первичных ресничках в клетках почечных канальцев и холангиоцитах.
Одиночная врожденная киста печени (непаразитарная) может возникнуть в детстве и в некоторых случаях м.б. выявлена при пренатальном УЗИ. Могут присутствовать вздутие живота и боль, может пальпироваться образование с нечеткими краями в правом верхнем квадранте живота.
Эти доброкачественные поражения лучше не беспокоить, если они не сдавливают соседние структуры или не возникает осложнений (напр., кровоизлияние в кисту). Оперативное лечение назначается пациентам с симптомами и растущими кистами.
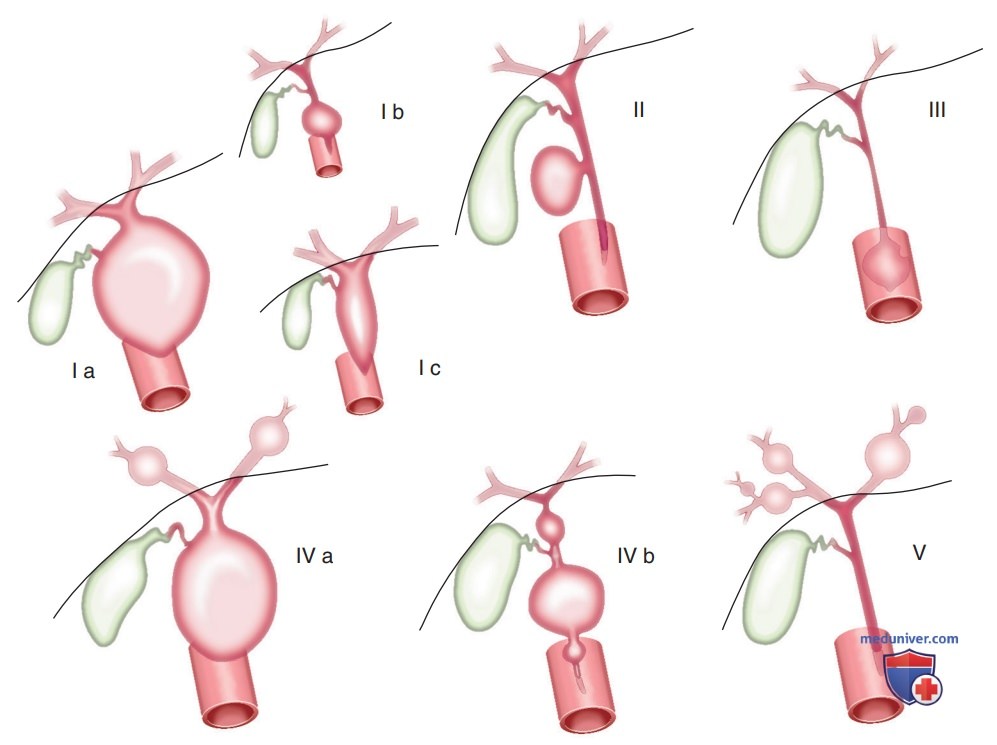
а) Кисты холедоха. Кисты холедоха — врожденные расширения общего желчного протока, которые могут вызвать прогрессирующую обструкцию ЖВП и билиарный цирроз. Цилиндрические (веретенообразные) и сферические (мешковидные) кисты внепеченочных протоков — наиболее распространенные типы (см. табл. 1). Кисты холедоха классифицируются по Тодани (рис. ниже). Кисты холедоха I типа — наиболее распространенный вариант.
Классификация кист холедоха по Тодани и соавт (Todani et al): Ia — наиболее распространенный тип; Ib — сегментарная дилатация; Ic — диффузная дилатация; II — дивертикул; III — холедохоцеле; IVa — множественные кисты (внутри — и внепеченочные); IVb — множественные кисты (внепеченочные); V — одиночное или множественное расширение в/печеночных протоков (болезнь Кароли).
Они включают мешковидное или веретенообразное расширение общего желчного протока. Кисты II типа — врожденные дивертикулы, выступающие из общего желчного протока. Кисты III типа или холедохоцеле включают грыжу в/дуоденального сегмента общего желчного протока в ДПК. Кисты IVa типа или болезнь Кароли включают множественные в/печеноч-ные и внепеченочные кисты. Кисты IVb типа поражают только внепеченочный проток. Одиночные кисты печени (тип V) встречаются очень редко.
Патогенез кист холедоха остается неизученным. Некоторые авторы предполагают, что соединение общего желчного протока и протока ПЖЖ до их входа в сфинктер Одди может привести к рефлюксу ферментов ПЖЖ в общий желчный проток. Это вызывает воспаление, локальное ослабление и расширение протока. Также высказано предположение, что врожденный стеноз дистальный сегмент желчного дерева приводит к повышению в/просветного давления и дилатации проксимальных отделов ЖВП.
Др. возможные варианты рассматривают кисты холедоха как пороки развития общего желчного протока или проявление инфекционного заболевания, включающего неонатальный гепатит и атрезию ЖВП.
Около 75% случаев проявляются в детстве. У младенцев развивается холестатическая желтуха. Если не устранить обструкцию ЖВП, то может быстро развиться тяжелая дисфункция печени, включая асцит и коагулопатию. Редко удается пропальпировать какое-либо образование. У детей старшего возраста классическая триада — боли в животе, желтуха и пальпируемое образование — встречается в <33% случаев.
Могут присутствовать признаки острого холангита (лихорадка, болезненность в правом подреберье, желтуха и лейкоцитоз). Диагноз ставится с помощью УЗИ. Использование этой методики позволяет выявлять кисты холедоха в/утробно. Магнитно-резонансная холангиография полезна при предоперационной оценке анатомии кисты холедоха.
Кисты холедоха могут перерождаться в холангиокарциному, поэтому предпочтительный метод лечения — первичное иссечение кисты и холедохоеюностомия методом Roux-en-Y. Послеоперационное течение может осложниться рецидивирующим холангитом или стриктурой в месте анастомоза. Необходимо длительное наблюдение, чтобы убедиться в отсутствии ЗНО.
б) Аутосомно-рецессивная поликистозная болезнь почек. ARPKD проявляется преимущественно в детском возрасте. Двустороннее увеличение почек вызвано генерализованным расширением собирательных канальцев. Это расстройство неизменно связано с врожденным фиброзом печени и различной степенью эктазии желчных протоков, которые подробно обсуждаются позже.
Ген поликистоза почек и печени 1 (PKHD1), мутированный при ARPKD, кодирует белок фиброцистин/полидуктин. Этот белок локализуется в ресничках на апикальном домене почечных собирательных клеток и холангиоцитов. Первичным дефектом при ARPKD м.б. цилиарная дисфункция, связанная с аномалией этого белка. Фиброцистин/полидуктин играет роль в регуляции клеточной адгезии, отталкивании и пролиферации или регуляции и поддержании почечных собирательных канальцев и желчных протоков.
Его точная роль в нормальном и кистозном эпителии остается неизвестной. Заболевания почек и печени независимы и различаются по тяжести. Они не объясняются типом мутации PKHD1.
Фенотипическая изменчивость среди пораженных болезнью братьев и сестер предполагает важность генов-модификаторов, а также влияние окружающей среды.
При ARPKD кисты возникают как эктатические расширения собирательных канальцев и желчных протоков, которые остаются в преемственности с их структурами происхождения. ARPKD проявляется в раннем возрасте, часто вскоре после рождения, протекает тяжелее, чем АуД-поликистозная болезнь почек (ADPKD). УЗИ может визуализировать большие почки, также описываемые как яркие, с низким содержанием или отсутствием околоплодных вод (маловодие). Но во многих случаях признаки ARPKD не визуализируются при сонографии до III триместра или после рождения.
Пациенты с ARPKD могут умереть в перинатальном периоде из-за почечной недостаточности или дисгенеза легких. Почки у этих пациентов заметно увеличены, и их функции нарушены. ДН м.б. результатом сдавления ГК сильно увеличенными почками, задержки жидкости или сопутствующей гипоплазией легких. В пределах семьи отмечена тенденция передавать клинические и патологические признаки потомству. Но наблюдаются некоторые различия в тяжести заболевания и времени его проявления в одной и той же семье. У пациентов, переживших младенчество из-за более мягкого фенотипа заболевания почек, патология печени может оказаться важной частью расстройства.
Заболевание печени при ARPKD сочетается с врожденным пороком развития печени с различной степенью перипортального фиброза, гиперплазией желчных протоков, эктазией и дисгенезом. Начальные симптомы вызваны поражением печени у 26% пациентов. Это может клинически проявляться как вариабельное кистозное расширение в/печеночного билиарного дерева с врожденным фиброзом печени. Врожденный фиброз печени и болезнь Кароли — результат нарушения ремоделирования эмбриональной протоковой пластинки печени.
Порок развития протоковой пластинки относится к сохранению избыточных эмбриональных структур желчных протоков в портальных трактах. Пациентам с ARPKD с рецидивирующим холангитом или осложнениями портальной гипертензии может потребоваться комбинированная трансплантация печени и почек.
в) Кистозное расширение внутрипеченочных протоков (болезнь Кароли/синдром Кароли). При болезни Кароли наблюдается изолированная эктазия или необструктивное сегментарное расширение более крупных в/печеночных протоков. Синдром Кароли — более распространенный вариант, при котором пороки развития мелких желчных протоков связаны с врожденным фиброзом печени. Врожденное мешковидное расширение может поражать несколько сегментов в/печеночных желчных протоков. Расширенные протоки выстланы кубовидным эпителием и находятся в непрерывной связи с основной системой протоков (вариант нормы).
Кисты холедоха также наблюдаются при болезни Кароли. Расширение желчных протоков приводит к застою желчи и образованию билиарного сладжа и в/протоковому литиазу. Существует выраженная предрасположенность к восходящему холангиту, который может усугубляться образованием камней в аномальных желчных протоках.
Больные испытывают симптомы острого холангита в детском или молодом возрасте. Возникает лихорадка, боль в животе, легкая желтуха и зуд, а также наблюдается слегка увеличенная, болезненная печень. Во время эпизодов острой инфекции может наблюдаться повышенная активность ЩФ, повышенный уровень прямого билирубина и лейкоцитоз. У пациентов с болезнью Кароли клинические признаки м.б. результатом сочетания повторяющихся приступов холангита, отражающих аномалии в/печеночных протоков и кровотечение из-за портальной гипертензии в результате фиброза печени.
УЗИ показывает расширение в/печеночных протоков, но окончательный диагноз и степень заболевания должны быть определены с помощью чрескожной чреспеченочной, эндоскопической или магнитно-резонансной холангиографии.
Холангит и сепсис следует лечить соответствующими АБ. При камнях может потребоваться хирургическое вмешательство. Частичная гепатэктомия может приводить к излечению в редких случаях, когда кистозное поражение ограничивается одной долей. В остальном прогноз остается сложным, в основном из-за трудностей в контроле холангита и билиарного литиаза и значительного риска развития холангиокарциномы.
г) Врожденный фиброз печени. Врожденный фиброз печени ассоциируется с ARPKD и морфологически характеризуется широкими зонами диффузного перипортального и перилобулярного фиброза. Эти зоны содержат искаженные структуры, подобные желчным протокам, часто сдавливающие или включающие центральные или сублобулярные вены (см. табл. 1). Островки паренхимы печени неправильной формы содержат нормального вида гепатоциты.
Болезнь Кароли и кисты холедоха связаны между собой. У большинства пациентов наблюдаются заболевания почек, в основном АуР-поликистозная болезнь почек и редко нефронофтизис. Врожденный фиброз печени также возникает как часть синдрома COACH (гипоплазия червеобразного отростка мозжечка, олигофрения, врожденная атаксия, колобома и фиброз печени). Врожденный фиброз печени описан у детей с врожденным нарушением гликозилирования, вызванным мутациями в гене, кодирующем фосфоманнозизомеразу.
Клинически определено несколько различных форм врожденного фиброза печени: в результате портальной гипертензии (наиболее частая), холангиогенная, смешанная и латентная. Расстройство начинается в детстве, с гепатоспленомегалии или кровотечения, вторичного по отношению к портальной гипертензии. В недавнем исследовании спленомегалия как маркер портальной гипертензии развилась в раннем возрасте и наблюдалась у 60% детей <5 лет. У таких пациентов может развиться холангит, поскольку у них встречаются аномалии ЖВП даже без болезни Кароли.
Гепатоцеллюлярная функция хорошо сохраняется. Активность аминотрансфераз в сыворотке крови и уровень билирубина обычно в норме при отсутствии холангита и холедохолитиаза. Активность ЩФ в сыворотке крови м.б. незначительно повышена. Уровень сывороточного альбумина и ПВ в норме. Биопсия печени редко требуется для постановки диагноза, особенно у пациентов с очевидным заболеванием почек.
Лечение этого расстройства должно быть сосредоточено на профилактике кровотечений из варикозно расширенных вен пищевода и агрессивном лечении холангита АБ. Редкие эпизоды легкой кровопотери м.б. устранены с помощью эндоскопической склеротерапии или перевязки варикозных вен. Портокавальный анастомоз может снизить портальную гипертензию после более сильного кровотечения. Прогноз м.б. значительно улучшен с помощью процедуры шунтирования, но выживаемость у некоторых пациентов ограничена почечной недостаточностью.
д) Аутосомно-доминантная поликистозная болезнь почек. ADPKD — наиболее частое наследуемое кистозное заболевание почек. Оно поражает 1:1000 родившихся живыми новорожденных. Характеризуется прогрессирующим развитием и увеличением кист почек, множеством внепочечных проявлений. Существует высокая степень в/семейной и межсемейной вариабельности клинических проявлений заболевания.
ADPKD вызвана мутацией в одном из двух генов, PKD1 (первый ген аутосомно-доминантной поликистозной болезни почек) или PKD2 (второй ген аутосомно-доминантной поликистозной болезни почек), на долю которых приходится 85-90% и 10-15% случаев. Белки полицистин-1 и полицистин-2, кодируемые этими генами, экспрессируются в клетках почечных канальцев и в холангиоцитах. Поликистин-1 функционирует как механосенсор в ресничках, обнаруживая движение жидкости по канальцам. Он передает сигнал через поликистин-2, который действует как кальциевый канал.
Чаще всего выявляются увеличенные неинфицированные кисты печени. Др. поражения печени, включая пороки развития протоковой пластинки, врожденный фиброз печени и микрогамартомы желчных путей (комплексы фон Мейенбурга), редко ассоциируются с ADPKD. У 50% пациентов с почечной недостаточностью наблюдаются явные кисты печени, которые происходят из желчных путей, но не связаны с ними. Кисты печени увеличиваются с возрастом. В одном исследовании распространенность кист печени составила 58% у пациентов 15-24 лет. Кистогенез печени зависит от эстрогенов.
Хотя частота кист одинакова у мужчин и женщин, развитие крупных кист печени считается осложнением терапии эстрогенами у женщин. Кисты печени часто протекают бессимптомно, но могут вызывать боль и иногда осложняются кровотечением, инфекцией, желтухой из-за сдавления желчных протоков, портальной гипертензией с варикозным кровотечением или затруднением оттока печеночных вен из-за механического сдавливания печеночных вен. Это приводит к небольшой гепатомегалии и экссудативному асциту. Может развиться холангиокарцинома. Субарахноидальное кровоизлияние м.б. результатом ассоциированных с кистами печени церебральных артериальных аневризм.
Отдельным пациентам с тяжелой симптоматической поликистозной болезнью печени и благоприятной анатомией приносит пользу резекция или фенестрация печени. Может потребоваться комбинированная трансплантация печени и почек. Известны значительные доказательства роли циклического АМФ в пролиферации эпителия и секреции жидкости при экспериментальной почечной и печеночной кистозной болезни. Несколько клинических испытаний у взрослых показали, что аналоги соматостатина могут замедлить расширение кисты печени за счет блокады секреции циклического АМФ, индуцированной секретином, и секреции жидкости холангиоцитами.
е) Аутосомно-доминантная поликистозная болезнь печени. АуД-поликистоз печени — генетическое заболевание, при котором развиваются множественные кисты, не связанные с кистозной болезнью почек. Кисты печени возникают из ЖВП, но не связаны с ними. Девочки болеют чаще, чем мальчики, и кисты увеличиваются во время беременности. Кисты редко выявляются в детском возрасте. Осложнения кист вызваны эффектами местного сдавления, инфекциями, кровотечением или разрывами. Гены PRKCSH и SEC63 связаны с АуД-поликистозом печени. Эти гены кодируют гепатоцистин и Sec63.
Гепатоцистин — субстрат протеинкиназы С adK-H, который участвует в правильном складывании и созревании гликопротеинов. Он локализован в эндоплазматической сети. SEC63 кодирует белок SEC63P — компонент белка механизма транслокации в эндоплазматическом ретикулуме.
Видео №1: УЗИ кисты холедоха (общего желчного протока) и УЗИ при болезни Кароли