АуД-ПБП, также известная как поликистоз почек у взрослых, является наиболее распространенным наследственным заболеванием почек с частотой 1:400-1000. Это системное заболевание с возможным образованием кист во многих органах (печень, ПЖЖ, селезенка, ГМ) и развитием мешковидных аневризм ГМ.
а) Патология. Обе почки увеличены и представлены большими кортикальными и медуллярными кистами, исходящими из всех областей нефрона.
б) Патогенез. Ок. 85% пациентов с АуД-ПБП имеют мутации, картируемые в гене PKD1 на коротком плече хромосомы 16, которая кодирует полицистин, трансмембранный гликопротеин. Еще 10-15% мутаций картируются в геном PKD2 на длинном плече хромосомы 4, которая кодирует полицистин 2, предполагаемый неселективный катионный канал.
Большинство мутаций, по-видимому, уникальны для конкретной семьи. В настоящее время мутация м.б. обнаружена у 85% пациентов с хорошо изученным заболеванием. Приблизительно 8-10% пациентов будут иметь мутации de novo, вызывающие данную патологию. Мутации PKD1 обусловливают развитие более тяжелой формы ПБП, нежели мутации PKD2.
Патофизиология заболевания, по-видимому, представлена нарушением нормальных мультимерных цистопротеиновых комплексов с последующей аномальной в/клеточной передачей сигналов, приводящей к аномальной пролиферации, канальцевой секреции и образованию кист.
Аномальная экспрессия фактора роста в сочетании с низким уровнем в/клеточного кальция и повышенным уровнем циклического АМФ, по-видимому, является важным фактором, ведущим к образованию кист и прогрессирующему увеличению почек. Сообщалось о мутациях в GANAB у PKD1-/PKD2-негативных пациентов.
в) Клиническая картина. Тяжесть заболевания почек и клинические проявления АуД-ПБП сильно различаются. Явная симптоматика чаще всего возникает на четвертом / пятом десятилетии жизни. Однако симптомы, включая макрогематурию, двустороннюю боль в области поясницы, объемные образования в животе, АГ и ИМП, могут наблюдаться у новорожденных, детей и подростков.
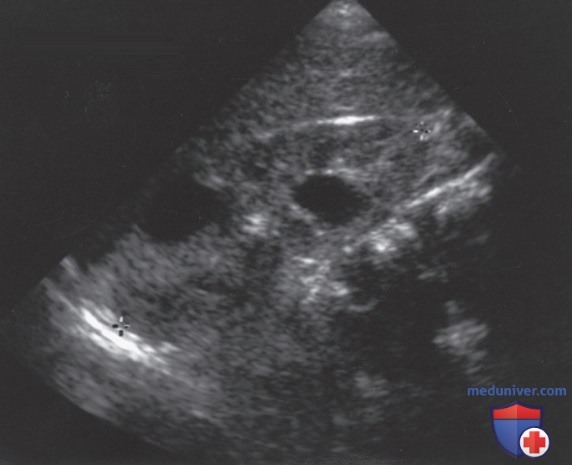
С увеличением доступности абдоминального УЗИ в педиатрии и в семьях с АуД-ПБП при скрининге детей из группы риска с возможным бессимптомным заболеванием (с принятием в США Закона о недискриминации генетической информации), большинству детей с АуД-ПБП диагноз устанавливается на основании аномальных результатов УЗИ почек при отсутствии симптомов. УЗИ почек обычно выявляет множественные двусторонние макрокисты в увеличенных почках (рис. ниже), хотя у детей на ранней стадии болезни почки м.б. нормальных размеров, а поражение м.б. одностороннем.
Ультразвуковое исследование мальчика 18 мес с аутосомно-доминантной поликистозной болезнью почек демонстрирует увеличение почек (10 см) и две большие кисты
АуД-ПБП — это полиорганная болезнь, поражающая многие типы тканей. Кисты могут существовать бессимптомно, локализуясь в печени, ПЖЖ, селезенке и яичниках, их наличие способствует постановке диагноза в детском возрасте. В/черепные аневризмы, которые, по-видимому, сегрегируют в определенных семьях, имеют общую распространенность 15% и являются важной причиной смертности у взрослых, но могут возникать и у детей.
ПМК наблюдается у ~12% детей; у взрослых отмечаются аневризмы аорты и КА, недостаточность аортального клапана. У детей могут формироваться грыжи, бронхоэктазы и дивертикулы кишечника.
г) Диагностика. АуД-ПБП подтверждается наличием увеличенных почек с двусторонними макроцистами при наличии родственника первой степени с такой же патологией. Мутации de novo встречаются у 8-10% пациентов с впервые диагностированным заболеванием.
Диагноз м.б. поставлен детям раньше, чем у родителя, что делает УЗИ почек у родителей важным диагностическим мероприятием, которое следует проводить в семьях без явного семейного анамнеза. Среди пациентов с генетически детерминированной АуД-ПБП результаты скринингового УЗИ почек м.б. в норме в <20% случаев к 20 годам и <5% случаев к 30 годам.
В семьях с известной АуД-ПБП пренатальный диагноз устанавливается по УЗИ при наличии увеличенных почек с/без кист. Пренатальный анализ ДНК проводится семьям с данной патологией, вызванной мутациями в генах PKD1 / PKD2.
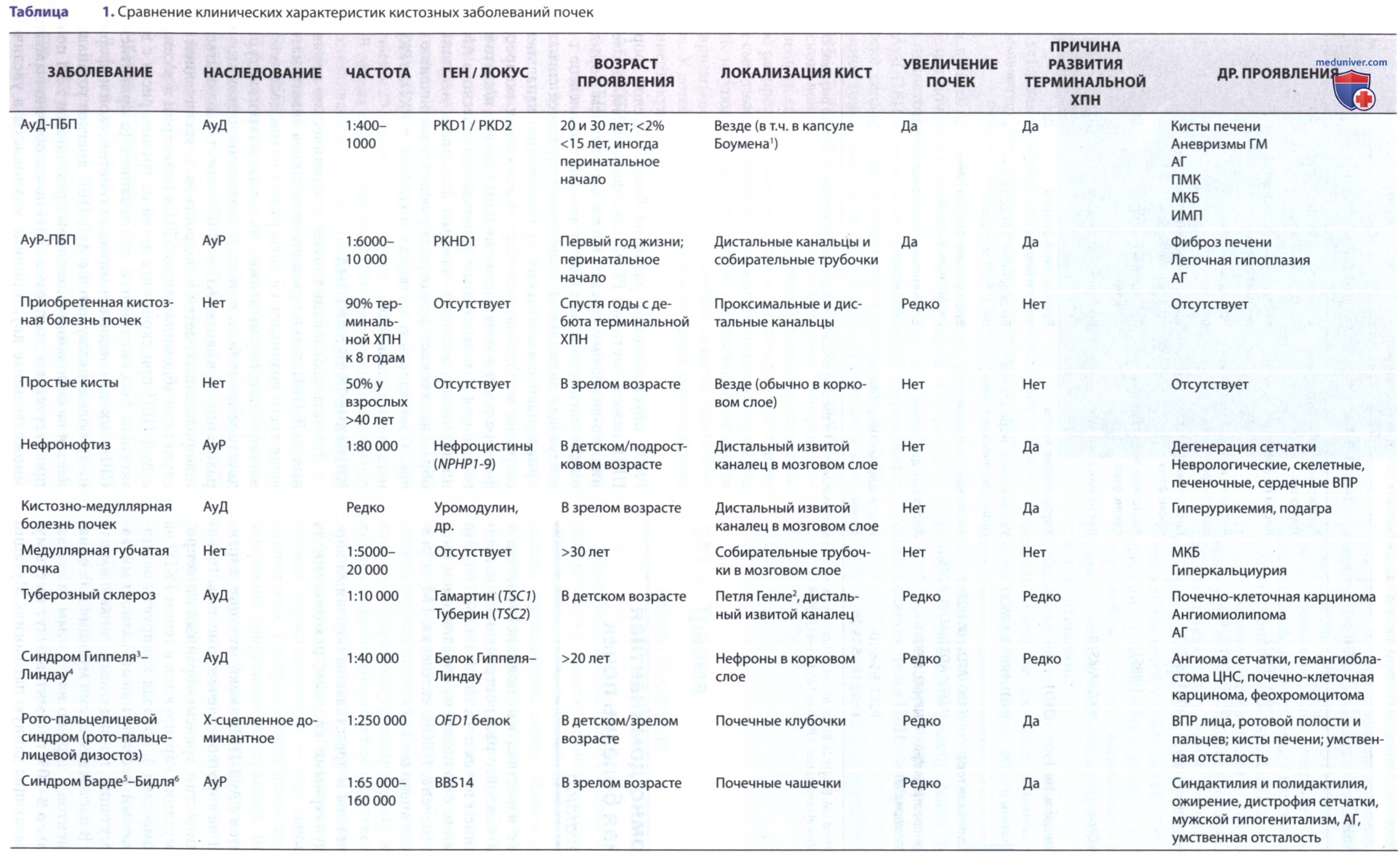
ДД включает кисты почек, развитие которых обусловлено гломерулокистозной болезнью почек, туберозным склерозом и болезнью Гиппеля-Линдау, имеющих АуД-тип наследования (см. табл. 1). Неонатальные проявления АуД-ПБП и АуР-ПБП спутать сложно.
д) Лечение и прогноз. Лечение АуД-ПБП в первую очередь поддерживающее. Контроль АД имеет решающее значение, т.к. скорость прогрессирования заболевания коррелирует с АГ. иАПФ и/или антагонисты рецепторов ангиотензина II являются ЛП выбора. Ожирение, избыток пищевой соли и белка, употребление кофеина, курение, многоплодная беременность и мужской пол, по-видимому, ускоряют прогрессирование заболевания. Пожилые пациенты с семейным анамнезом разрыва в/черепной аневризмы должны быть обследованы на предмет церебральных аневризм.
Хотя неонатальная АуД-ПБП м.б. фатальной, долгосрочное выживание пациента и сохранность почек возможны у детей, переживших неонатальный период. АуД-ПБП, возникающая у детей старшего возраста, имеет благоприятный прогноз, при этом нормальная функция почек наблюдается у >80% детей. Боль м.б. проявлением инфекции, кровоизлияния, разрыва кисты, конкрементов и опухолей, и ее следует надлежащим образом контролировать с помощью обезболивающих ЛП, в т.ч. в зависимости от ее этиологии.
Несмотря на то, что специфическая терапия для заболевания еще не доступна, клинические испытания на основе многообещающих доклинических лабораторных исследований продолжаются. Эти потенциальные методы лечения включают блокаторы ренин-ангиотензина, антагонистов V2-рецептора вазопрессина (толваптан) и аналоги соматостатина. Ценным ресурсом для пациентов и их семей является Фонд ПБП.