Наследуемые рецессивные спиноцеребеллярные атаксии: атаксия Фридрейха и другие
а) Атаксия Фридрейха. Атаксия Фридрейха — наиболее четко описанная и часто встречающаяся спиноцеребеллярная дегенерация. Частота встречаемости гена составляет 1:110 человек в Англии (Harding 1981a), и примерно один из 10000 человек в Швеции имеет клинические проявления.
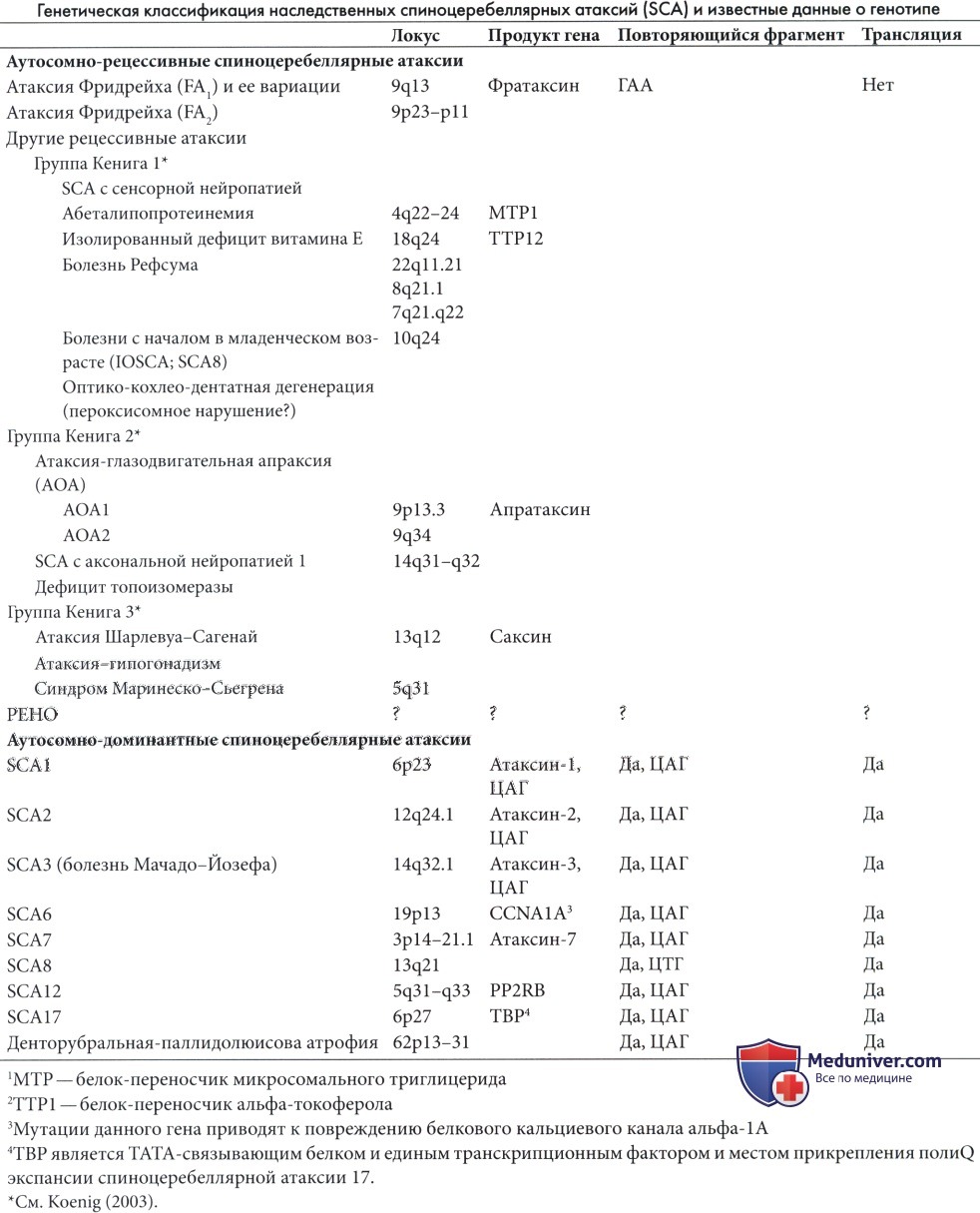
Ген атаксии Фридрейха включает повторы ГА А последовательности в интроне 1, который распространен у пациентов (120-1700 повторов). Продуктом нормального гена является белок фратаксин, функция которого не полностью ясна. 94% пациентов с типичной атаксией Фридрейха являются гомозиготами по ГАА экспансии, тем не менее продолжительность повтора на каждой хромосоме из пары неодинакова (Durr et al., 1996a).
В редких случаях отмечается только одна мутация, но в такой ситуации выявляется точечная мутация в гомозиготном локусе (Campuzano et al., 1996). Выраженная длина повтора коррелирует с началом в раннем возрасте, более стремительным течением и наличием кардиомиопатии (Durr et al., 1996a).
Второй ген на хромосоме 9p23-p11 является причиной редких случаев (Фридрейха 2), клинически нечетко отличаемых от 1 типа (Christodoulou et al., 2001).
Основным патологическим проявлением является дистальная аксональная нейропатия, которая поражает нейроны длинных восходящих и нисходящих трактов спинного мозга и крупные сенсорные волокна периферических нервов и ганглии задних корешков (Said et al., 1986). Также зарегистрирована утрата нервных волокон в зрительных путях, а мозжечок остается непораженным.
Сердце увеличено, и более чем в половине случаев отмечается гипертрофическая кардиомиопатия с некрозом волокон и фиброзом, преимущественно затрагивающим левый желудочек.
Критерии диагностики атаксии Фридрейха (Harding, 1981a) включают начало до 25 лет (обычно до 16 лет), аутосомно-рецессивное наследование и сочетанное поражение крупных сенсорных волокон периферических нервов, мозжечкового тракта, пирамидного тракта и задних столбов.
Тем не менее, степень фенотипической вариабельности велика, в некоторых случаях отмечается позднее начало и/или меньшая выраженность симптомов и вариабельное течение, и некоторые пациенты прикованы к инвалидной коляске в раннем подростковом возрасте, в то время как другие способны самостоятельно передвигаться почти до 40 лет (Montermini et al., 1997).
По неофициальным данным, к доминантным случаям относится большая часть наследственной моторной и сенсорной нейропатии со скелетными деформациями и утратой чувствительности, но некоторые случаи не поддаются классификации.
Клинические проявления атаксии Фридрейха были описаны у 115 пациентов из 90 семей (Harding, 1981b). Основные проявления представлены в таблице ниже.
Заболевание чаще всего начинается в возрасте 5-16 лет, в редких случаях — в возрасте 2-5 лет. Прогрессирующая атаксия нижних конечностей с нарушением походки является основным проявлением, в то время как поражение верхних конечностей, приводящее к неуклюжести, в ранние сроки отмечается только в 25% случаев.
Сколиоз, тремор и изменения со стороны сердца редко являются первыми проявлениями, но формируются со временем, особенно при раннем начале заболевания. Конская стопа также является ранним симптомом. При осмотре в 70-95% случаев выявляется отсутствие глубоких рефлексов.
Дизартрия, пирамидные знаки со стороны ног и утрата глубокой и вибрационной чувствительности могут появляться позже, но относятся к постоянным симптомам. Нистагм встречается нечасто (20% случаев), медленные изломанные следящие движения глаз выявляются в 12% случаев (Harding, 1981b). Нестабильность фиксации является типичным проявлением (Alper и Narayanan, 2003).
Нередко встречается атрофия зрительного нерва, а глухота отмечается только у 10% пациентов. Дистальная атрофия отмечается практически в половине случаев. Интеллект не страдает.
Поражение сердца по результатам ЭКГ обнаруживается в трети случаев и даже чаще, если ЭКГ проводится систематически. Изменения зубца Т и аномалии сегмента ST являются ранними признаками сердечной недостаточности и единственным ее проявлением.
В конечном счете формируется прогрессирующая сердечная недостаточность или аритмия с фибрилляцией предсердий, половина пациентов умирает от сердечной недостаточности (Leone et al., 1988).
Течение заболевания медленное, но прогрессирующее. В среднем пациенты утрачивали способность ходить к 25 годам, со средней продолжительностью заболевания 15,5 лет.
Сахарный диабет является дальнейшим осложнением и развивается у 10% пациентов. Он имеет тенденцию сочетаться с атрофией зрительного нерва, в некоторых случаях диабетическая кома является причиной смерти.
Атипичные формы включают легкие случаи, которые, возможно, связаны с одним и тем же локусом 9-й хромосомы. К данной группе относятся случаи сохранения сухожильных рефлексов (Palau et al., 1995) и поздние формы с началом в раннем взрослом возрасте (De Michele et al., 1994).
Результаты одной из недавних работ, в которой использовалось возможное выявление мутантного гена, предполагают, что клиническая картина более вариабельна, чем считалось раньше (Palau et al., 1995; Pandolfo 2003). 25% пациентов в рамкам одного крупного исследования имели одно или более атипичное проявление (начало после 25 лет, сохранение или даже оживление сухожильных рефлексов или отсутствие симптома Бабинского).
Возраст начала превышал 20 лет у 19 из 114 пациентов (De Michele et al., 1994). Сохраненные рефлексы среди пациентов, которые в остальном соответствуют всем критериям, зарегистрированы у значимого числа пациентов (Palau et al., 1995). Большая часть случаев, ранее отнесенных к рано начинающейся атаксии с сохранением глубоких сухожильных рефлексов (состояния, отличного от атаксии Фридрейха) (Harding, 1981b; Klockgether et al., 1991), по результатам молекулярно-генетических исследований, вероятно, имеют отношение к болезни Фридрейха.
Эта группа, очевидно, была гетерогенной как при раннем (<3 лет), так и позднем начале, поэтому нозологическая ситуация в данной группе не совсем ясна, хотя атаксия Фридрейха по всей вероятности относилась к большинству случаев.
Данное обстоятельство может касаться также случаев начала заболевания в детском возрасте, описанных как семейная спастическая атаксия с рецессивным типом наследования.
Акадский тип, отмечаемый в специфической популяции Луизианы, характеризующийся более легким течением и меньшей частотой кардиомиопатии, при анализе групп сцепления был также расценен как имеющий общее происхождение с классической атаксией Фридрейха.
Диагноз атаксии Фридрейха основан на клинических изменениях. КТ обычно не отличается от нормы, что исключает опухоли или гидроцефалию. На МРТ выявляется отсутствие атрофии мозжечка и истончения спинного мозга в шейном отделе (Wullner et al., 1993).
Скорость проводимости по двигательным и чувствительным нервам не отличается от нормы или немного замедлена, но вызванные сенсорные потенциалы отсутствуют или амплитуда их значимо снижена.
Вызванные зрительные потенциалы не соответствуют норме у двух третей пациентов (Pinto et al, 1988), световое зрение нарушается у 40% пациентов даже в случае нормальной электроретинограммы. Диагноз может быть подтвержден молекулярным генетическим исследованием.
Атаксию Фридрейха иногда сложно дифференцировать от других спиноцеребеллярных дегенераций, описанных далее, в особенности от атипичных и неполных форм, которые нередко встречаются на начальных стадиях заболевания. Атаксия-телеангиэктазия характеризуется более ранним началом. Заподозрить диагноз можно на основании кожных проявлений и рецидивирующих инфекций.
Недавно описанное состояние атаксии-зрительной моторной апраксии I типа характеризуется изменениями движений глаз. Так называемые промежуточные формы между атаксией Фридрейха и наследственной дегенеративной моторной и сенсорной нейропатией обычно можно отнести к обоим типам. Синдром Русси-Леви не является неполной формой атаксии Фридрейха, а относится к вариантам наследственных дегенеративных гипертрофических нейропатий.
Неврологические формы наследственной моторной и сенсорной нейропатии относятся к самым частым случаям ошибочных диагнозов. Важно не путать их с атаксией Фридрейха, так как они характеризуются более благоприятным прогнозом. Первичная и вторичная недостаточность витамина Е может симулировать атаксию Фридрейха (Ben Hamida et al., 1993) и поддается лечению.
До недавнего времени лечение атаксии Фридрейха отсутствовало за исключением лечебной физкультуры и поддержания максимальной возможной активности. Хирургическое лечение может продлить способность ходить. Недавние работы, касающиеся роли окислительного стресса и его предотвращения, дают определенную надежду.
Фратаксин, продукт гена Фридрейха, играет важную роль в сборке комплекса железосерного белка, необходимого для митохондриального контроля железа и, возможно, обладающего функциями антиоксиданта (Bradley et al; 2004, Rustin et al, 2004; Hart et al., 2005). Правда, действительная роль митохондриального железа и окислительного стресса вызывает сомнения (Seznec et al., 2004, 2005). Тем не менее, активно продолжаются исследования с применением хелаторов железа и антиоксидантов (Hart et al., 2005).
Кофермент Q и витамин Е могут оказывать некоторый положительный эффект (Boddaert et al., 2007). Продемонстрировано, что идебенон (аналог кофермента Q) может приводить к отсрочке или уменьшать выраженность кардиомиопатии и оказывать некоторое действие на атаксию (Seznec et al., 2005). В настоящее время терапевтические стратегии обсуждаются (Schols et al., 2004).
б) Другие рецессивно наследуемые спиноцеребеллярные атаксии. Данные заболевания являются наиболее редкими (Di Donato et al., 2001). Кениг (2003) разделил их на три подгруппы:
1) заболевания, при которых есть данные о поражении спиноцеребеллярой системы и сенсорной нейропатии,
2) заболевания, при которых сенсомоторная нейропатия является основным признаком,
3) заболевания, при которых отмечается практически изолированное поражение мозжечка.
Данные заболевания представлены в отдельной статье на сайте. Представляющие особый интерес болезни этой группы описаны ниже.
Изолированный дефицит витамина Е представляет терапевтический интерес, так как лечение может стабилизировать или даже уменьшать выраженность клинических проявлений (Roubertie et al., 2003; Mariotti et al., 2004). Тем не менее, лечение не полностью предотвращает поздние проявления неврологических изменений, несмотря на первичный и стойкий эффект в отношении нейропатии. Абеталипопротеинемия и гипобеталипопротеинемия описаны в отдельной статье на сайте.
Спиноцеребеллярная атаксия с началом в младенческом возрасте (IOSCA), так же известная как SCA8, связана с мутацией 10-й хромосомы. Данное заболевание регистрируется преимущественно в Финляндии как синдром О-НАНА (офтальмоплегия, гипоакузия, атаксия, гипотония и атетоз). Заболевание характеризуется скорее ассоциативными клиническими проявлениями. Начало в периоде новорожденности или в течение первого года жизни (Lonnqvist et al., 1998). Течение прогрессирующее. Сенсоневральная тугоухость является основным симптомом и может проявляться резко, часто после трехлетнего возраста, результатом чего является утрата речи.
Синдром Боше-Нойхаузера характеризуется гипогонадотропным гипогонадизмом в сочетании со спиноцеребеллярным синдромом и пигментной ретинопатией (Rump et al., 1997). Оптико-кохлео-дентатный синдром является очень редким заболеванием; ранняя его форма с гипотонией с рождения, слепотой, глухотой и ранней смертью недавно была отнесена к пероксисомным заболеваниям (Ferrer et al., 1987; Schroder et al., 2004).
Вторая подгруппа Кенига включает атаксию со зрительной моторной апраксией (АОА), которая делится на два типа: AOA1 является одной из наиболее распространенных форм детского возраста и описана вместе с атаксией-телеангиэктазией, несмотря на то, что ее физиология кажется более сходной с спиноцеребеллярными атаксиями (SCA, см. далее). Одним из важных биологических признаков является гипоальбуминемия, которая практически постоянно обнаруживается и имеет диагностическую значимость. АОА 2 типа встречается реже и начинается позже (в позднем подростковом или раннем взрослом возрасте). Умеренно повышенный уровень альфа-фетопротеина отмечается в 75% случаев.
Клинические проявления AOA1 очень напоминают проявления атаксии-телеангиэктазии, но без признаков экстраневрологических поражений.
В отличии от атаксии-телеангиэктазии, AOA1 не связана с повышением уровня альфа-фетопротеина, хромосомными аномалиями, склонностью к раковым опухолям или повышенной радиочувствительностью культуры фибробластов (Le Ber et al., 2005). Редким, но интересным состоянием является спиноцеребеллярная атаксия с аксональной нейропатией 1 (SCA1), которая фактически является нарушением репарации ДНК, вызванной отсутствием фермента топоизомеразы-фосфодиэстеразы-1 (TDP1) (E1-Khamisy et al., 2005).
В третьей подгруппе Кенига четко описана атаксия Шарлевуа-Сагеней. Изначально синдром был описан в Квебеке, но с тех пор регистрировался и в других частях света (Gucuyener et al., 2001). Заболевание связано с мутацией гена сакцина на 13-й хромосоме (Engert et al., 2000). Фенотипические проявления включают заметную спастичность и постоянное наличие полос на глазном дне с преобладанием миелиновых волокон, радиально расходящихся от диска зрительного нерва.
Два редких аутосомно-рецессивных синдрома включают очень медленно прогрессирующую атаксию и могут рассматриваться вместе с SCA. Несмотря на то, что патология и механизмы заболеваний отличаются, они проявляются несколькими общими клиническими симптомами.
Синдром Маринеску-Шегрена включает атрофию мозжечка, преимущественно затрагивающую червь, раннее начало медленно прогрессирующей атаксии, катаракту, легкую задержку умственного развития, иногда гипогонадизм (Sewry et al., 1988) и позднее развитие специфической миопатии (Superneau et al., 1987). Заболевание развивается в результате мутации гена SLI1, кодирующего белок-шаперон, ключевой регулятор основных функций эндоплазматической сети.
РЕНО синдром (прогрессирующая энцефалопатия с периферическими отеками, гипсаритмией и атрофией зрительного нерва, также называемая церебелло-оптический синдром) является рецессивным заболеванием, зарегистрированным преимущественно в Финляндии (Salonen et al., 1991), хотя регистрировались случаи и в других странах. Основными проявлениями являются рано начинающиеся припадки, легкий дисморфизм, периферические отеки и регрессия, начинающаяся в возрасте 3-5 месяцев. Атрофия зрительного нерва развивается к концу первого года (Haltia и Somer, 1993).