Глобоидно-клеточная лейкодистрофия (GLD, болезнь Краббе) является аутосомно-рецессивным заболеванием, вызванным дефицитом галактоцереброзид-бета-галактозидазы (галактоцереброзидазы) (Suzuki et al., 1971). Ген находится на хромосоме 14q24—32.1. Галактоцереброзиды являются наиболее специфичными липидами миелина головного мозга; их концентрация отражает созревание. Выделяют три подтипа GLD: наиболее распространенную младенческую форму и редкие формы (ювенильную и взрослую).
Нейропатология болезни Краббе характеризуется недостаточностью олигодендроцитов, отсутствием миелина и пролиферацией глиальных клеток в пораженных участках в сочетании с мононуклеарными эпителиоидными клетками и скоплениями крупных многоядерных глобоидных клеток. Поражение периферической нервной системы встречается часто, но может быть выражено не так сильно, как при MLD. Дефицит галактоцереброзид-бета-галактозидазы приводит к накоплению цереброзидов и психозина (галактозил сфингозина). Многие симптомы, отмечаемые при GLD, связаны с повышением уровня психозина, которые оказывает токсическое действие (Suzuki, 2003).
Наиболее распространенная младенческая форма манифестирует обычно в виде гипотонии, развивающейся до шестимесячного возраста. Кроме того, характерными симптомами являются чрезвычайная возбудимость, особенно заметны вздрагивание при громких звуках и прогрессирующая ригидность. При осмотре выявляется повышение мышечного тонуса и пирамидные знаки, а также опистотонусные судороги с ретракцией головы, часто вызываемые внешним воздействием.
Часто отмечаются судороги, которые могут представлять собой инфантильные спазмы с атипичным гипсаритмическим ЭЭГ-паттерном. Отсутствие глубоких сухожильных рефлексов часто контрастирует со спастичностью. Развивается атрофия зрительного нерва и слепота. Болезнь прогрессирует стремительно и смерть обычно наступает в возрасте до двух лет. Выделены клинические стадии болезни Краббе (Hagberg et al., 1969).
Во всех случаях инфантильной формы заболевания отмечается высокий уровень белка (более 70 мг/дл) в спинномозговой жидкости, а скорость проводимости нервов снижена. При формах с поздним началом уровень белка в спинномозговой жидкости может не отличаться от нормы. На МРТ в области белого вещества мозжечка выявляется снижение плотности в режиме Т1 и повышение плотности в режиме Т2. При МР-спектроскопии в пораженном белом веществе выявляется заметное повышение уровня инозит и холин-содержащих веществ, что отражает демиелинизацию и глиальную пролиферацию, сопровождающуюся снижением N-аспартил ацетата, что является признаком утраты нейронов (Brockmann et al., 2003а).
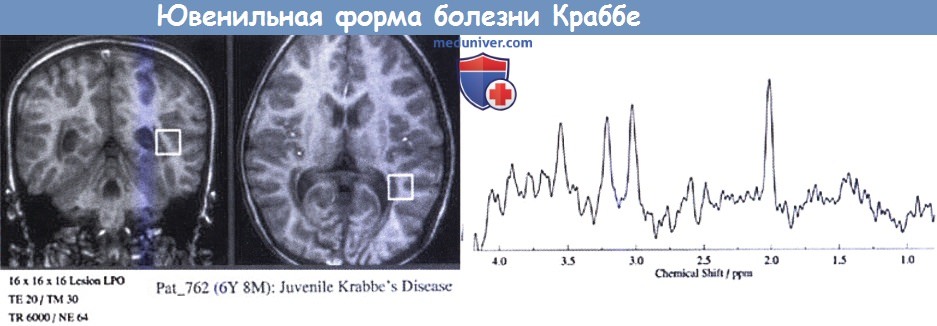
Ювенильная форма болезни Краббе.
На МРТ (вверху) видны только небольшие аномалии сигнала белого вещества.
Заметен низкий сигнал в области таламуса.
На МР-спектроскопии (внизу) видны высокие пики холина и миоинозитола и низкие лики N-ацетиласпартовой кислоты.
Диагностика GLD основана на характерных клинических симптомах, повышении уровня белка в спинномозговой жидкости и изменениях на МРТ. При позднем начале заболевания ключевые симптомы менее характерны и включают трудности при ходьбе, снижение зрения и трудности в обучении. При любой лейкодистрофии в сочетании с легкой периферической нейропатией следует подозревать GLD. Диагноз подтверждается путем оценки уровня галактоцероброзид-бета-галактозидазы в лейкоцитах и фибробластах.
Молекулярный анализ может выявить одну из более 60 мутаций, описанных при болезни Краббе (Kleiyer et al., 1997; Wenger et al., 1997).
Лечение имеет симптоматический характер. Бензодиазепины в высоких дозах могут быть эффективны для устранения чрезвычайной возбудимости и опистотонуса у некоторых пациентов. Предпринимались попытки трансплантации стволовых гемопоэтических клеток, но полученные результаты неоднозначны (Krivit et al., 1998). В последнее время восстановление нормального уровня галактоцереброзида в крови было достигнуто у 11 новорожденных с бессимптомным течением заболевания и 14 младенцев с симптомами болезни Краббе; результаты были достигнуты благодаря трансплантации пуповинной крови неродственных доноров (Escolar et al, 2005).
У младенцев, которым трансплантация была проведена до появления симптомов, в течение всего периода наблюдения (до трех лет) отмечалось прогрессирование центральной миелинизации, развитие и приобретение навыков в соответствии с возрастом. Трансплантация, проведенная после появления симптомов, не приводила к стойкому улучшению.
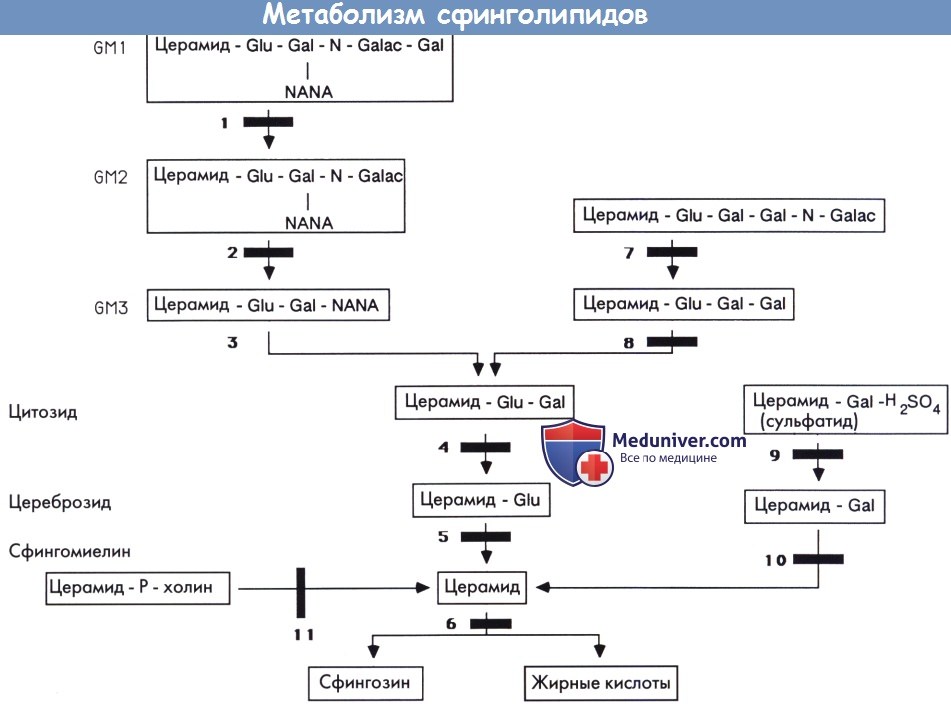
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.