Метахроматическая лейкодистрофия (MLD) впервые была описана Scholz и Greenfield. Scholz выявил генетическое происхождение заболевания и выделил его из группы болезней, описываемых как диффузный склероз. Типичные признаки в виде метахромазии при окрашивании крезил-фиолетом или толуидиновым синим в кислом растворе отделяют заболевание от так называемых ортохроматических или суданофильных лейкодистрофий. Окрашивание специфично выявляет сульфатиды, присутствующие не только в тканях головного мозга, но и в нервах и множестве соматических тканей, таких как почки, печень и яичники, а также в моче. Кроме метахромазии в периферических нервах также отмечается сегментарная демиелинизация.
Выявление метахроматического материала в эпителиальных клетках в моче до сих пор является полезным диагностическим тестом.
Метахроматическая лейкодистрофия (MLD) является аутосомно-рецессивным заболеванием, по оценкам с частотой 1 на 40000. Причиной заболевания является дефицит арилсульфатазы A (ARSA). Дефицит ARSA приводит к накоплению цереброзид сульфата (сульфатида). Генетический дефект локализован на 22 хромосоме (Polten et al., 1991). Накопление сульфатидов приводит к повреждению центрального и периферического миелина. Концентрация сульфатидов в пораженных тканях повышается, а концентрация цереброзида снижается.
Кроме аллелей ARSA (частота 0,5%), являющихся причиной MLD, существуют другие ARSA-аллели (частота 7-15%), называемые псевдодефицитным ARSA-геном (ARSA-PD) (Gieselmann et al., 1991). У 10-20% гомозигот по данному аллелю отмечается сниженная активность ARSA, но клинические симптомы и сульфатидурия не развиваются. Гомозиготность по ARSA-PD часто встречается в общей популяции; пациентам, у которых причиной неврологических симптомов является не ARSA-PD, может быть ошибочно установлен диагноз MLD. Псевдодефицитный аллель может быть выявлен непосредственно при исследовании ДНК. Кроме того, в редких случаях MLD может быть вызван мутациями, затрагивающими активатор белка (сапонин В), кодируемый геном на участке 10q21—22. Активатор белка активирует не только гидролиз сульфатидов под действием ARSA, но и гидролиз GM1-ганглиозидов.
Дефицит сапонина В может приводить к ювенильной или (в редких случаях) поздней младенческой форме MLD, но гистологически также отмечается накопление ганглиозидов. Диагностика проводился в культуре фибробластов: патологический метаболизм может быть восстановлен активатором белка (Gieselmann, 2003).
В соответствии с возрастом начала заболевания можно выделить три клинических типа MLD: поздний младенческий (40%), ювенильный (40%) и взрослый (20%). Поздняя младенческая форма является наиболее гомогенной и возникает в возрасте от шести месяцев до двух лет. У детей отмечается задержка развития или утрата способности к ходьбе: некоторые дети не способны ходить самостоятельно. Походка характеризуется спастичностью и атаксией. Может отсутствовать ахиллов рефлекс в сочетании с наличием двустороннего рефлекса Бабинского. В дальнейшем течении заболевания в клинической картине преобладают атрофия зрительного нерва и более генерализованная спастичность (MacFaul et al., 1982). Прогноз неблагоприятный, смерть наступает до 10 лет.
Время появления симптомов при ювенильной форме варьирует, заболевание может начинаться в возрасте 4-6 лет. Наблюдается нарушение походки в сочетании с трудностями обучения и поведенческими проблемами. Двигательные симптомы включают мозжечковые и пирамидные нарушения, а также признаки периферической нейропатии. Припадки и деменция являются поздними проявлениями. В случае начала заболевания во взрослом возрасте в клинической картине преобладают нарушения походки, экстрапирамидные симптомы и психиатрические отклонения.
Метахроматическую лейкодистрофию (MLD) следует подозревать у всех детей с прогрессирующими спастическо-атаксическими симптомами и парадоксальным отсутствием ахиллова рефлекса при наличии рефлекса Бабинского. При осмотре глазного дна может выявляться атрофия зрительного нерва. Показатели ЭЭГ обычно не отличаются от нормы или позволяют выявить некоторую медленную активность и эпилептиформные разряды на поздних стадиях заболевания. Электроретинограмма сохраняется неизмененной. Скорость проводимости чувствительных и двигательных нервов снижена. Практически всегда выявляется повышенное содержание белка в спинномозговой жидкости.
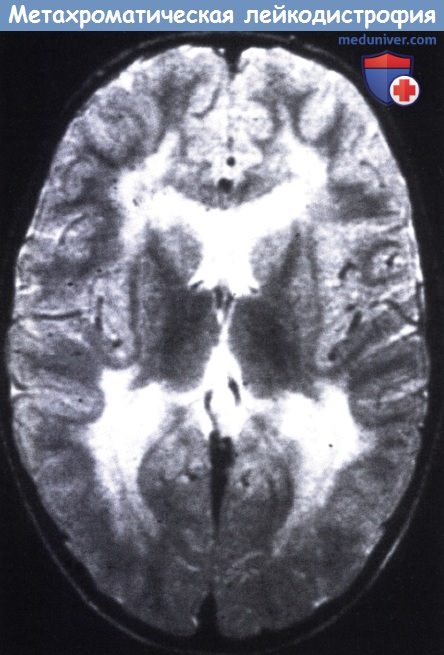
При всех трех типах заболевания на КТ и МРТ отмечаются изменения перивентрикулярного белого вещества с преобладанием поражения в лобной области. При ювенильной и взрослой формах заболевания U-волокна сохраняются, отмечается раннее поражение мозолистого тела, в пирамидном тракте могут выявляться признаки демиелинизации. Диагноз подтверждается на основании определения уровня ARSA и метахроматического материала в моче.
В настоящее время эффективные методы лечения отсутствуют. Предпринимались попытки трансплантации костного мозга или стволовых клеток гемопоэза, но полученные результаты имели противоречивый характер. В последнее время поднят вопрос о поддерживающей терапии арилсульфатазой-А (Matzner и Gieselmann, 2005).