Болезнь Краббе у детей - глобоидноклеточная лейкодистрофия
Болезнь Краббе — редкое нейродегенеративное заболевание с аутосомно-рецессивным типом наследования, выраженным распадом миелина и глобоидными тельцами в белом веществе мозга. Ген, определяющий развитие болезни Краббе (GALC), локализован на хромосоме 14q-24.3-q32.1. В основе заболевания лежит выраженный дефицит лизосомного фермента галактоцереброзид-b-галактозидазы, который отщепляет часть молекулы галактозы от церамидной порции галактоцереброзида. Болезнь Краббе — заболевание с деструкцией миелина, а не с формированием аномального миелина.
В норме миелинизация начинается в III триместре беременности, что соответствует быстрому увеличению активности галактоцереброзид-b-галактозидазы в головном мозге. У пациентов с болезнью Краббе метаболизм галактоцереброзида в процессе нормального обмена миелина невозможен в связи с дефицитом галактоцереброзид-b-галактозидазы. При введении галактоцереброзида в мозг экспериментальных животных наблюдается глобоидноклеточная реакция. Установлено, что сходный феномен имеет место и у людей: неметаболизирующийся галактоцереброзид стимулирует образование глобоидных клеток, что отражает деструкцию клеток олигодендроглии.
Поскольку олигодендроглиальные клетки отвечают за выработку миелина, гибель этих клеток приводит к распаду миелина, в результате которого образуется дополнительное количество галактоцереброзида; таким образом, формируется порочный круг деструкции миелина.
Симптомы болезни Краббе проявляются в первые несколько месяцев жизни и включают чрезмерную раздражительность и частый плач, необъяснимые эпизоды гиперпирексии, нарушение питания, рвоту и задержку прибавки массы тела. На первой стадии заболевания у детей часто диагностируется колика или аллергия на молоко, проводится соответствующее лечение с частым изменением формулы молочных смесей.
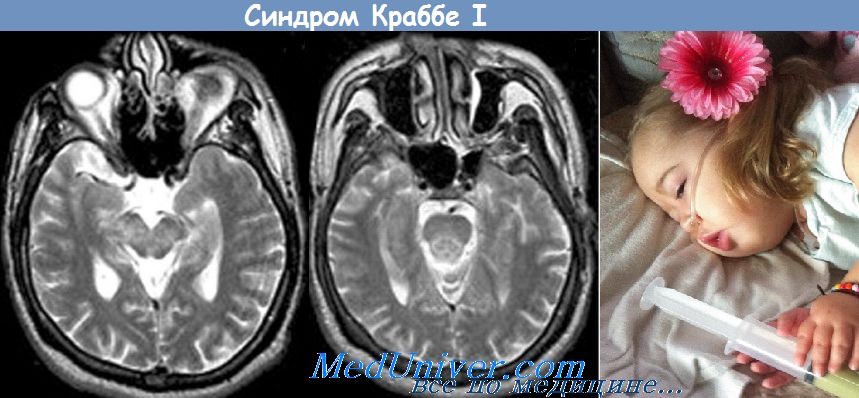
Генерализованные судороги могут возникать на ранней стадии. Изменение мышечного тонуса с развитием ригидности и опистотонуса, отсутствие зрительного внимания вследствие атрофии зрительных нервов появляются по мере прогрессирования заболевания. В поздней стадии основу клинической картины составляют такие симптомы, как слепота, глухота, отсутствие глубоких сухожильных рефлексов и децеребрационная ригидность. На КТ головного мозга без контрастирования возможно симметричное повышение плотности в области хвостатых ядер и таламуса. Большинство пациентов умирают к 2-летнему возрасту.
Описана болезнь Краббе с поздним дебютом — в школьном или подростковом возрасте. У пациентов с этим заболеванием выявляется атрофия зрительных нервов и корковая слепота, нередко в этих случаях ошибочно диагностируется АЛД. Медленно прогрессируют нарушение походки, спастичность и атаксия. Как и при классической болезни Краббе, в белом веществе большое количество глобоидных клеток, в лейкоцитах определяется дефицит галактоцереброзид-и-галактозидазы. В СМЖ повышено содержание белка, значительно снижена скорость проведения возбуждения по нервам вследствие сегментарной демиелинизации периферических нервов.
Амплитуда ЗВП постепенно уменьшается, и на поздних стадиях заболевания зрительный ответ отсутствует. При исследовании слуховых ВП ствола характерны только волны I и II. На КТ и МРТ выраженное уменьшение количества белого вещества, особенно в мозжечке и полуовальном центре (centrum semiovale), в сочетании с уменьшением количества (обеднением) субкортикальных U-волокон. Разработан метод пренатальной диагностики, основанный на определении активности галактоцереброзид-b-галактозидазы в ворсинах хориона или в культуре амниотических клеток.