Болезнь Александера (AXD) является первым описанным заболеванием, вызванным преимущественно нарушением функции астроцитов (Hanefeld, 2004; Mignot et al., 2004). Болезнь впервые была описана Александером в 1949 г. у 18-месячного мальчика с макроцефалией.
Нейропатологический признак заболевания представлял собой «палочковидные фусцинофильные тельца» в белом веществе головного мозга, которые в дальнейшем были идентифицированы как волокна Розенталя. Глиальный фибриллярный кислый белок (GFAP) присутствует в волокнах Розенталя в высокой концентрации и используется в качестве гистологического маркера.
Мутации гена GFAP (хромосома 17q21) у пациентов с болезнью Александера в дальнейшем были идентифицированы как причина аутосомно-рецессивного заболевания (Brenner et al., 2001).
В соответствии с клинической картиной выделяют три различных типа болезни Александера: младенческий, ювенильный и взрослый.
Наиболее распространенный младенческий тип относится к группе макроцефалических болезней белого вещества. У больных детей отмечается нормальный размер головы при рождении, а затем развивается медленно прогрессирующая мегалэнцефалия в сочетании со спастичностью, раздражимостью и эпилептическими припадками (Pridmore et al, 1993).
Острые эпизоды внутричерепной гипертензии часто отмечаются после легкой травмы головы. Смерть может наступить в возрасте до 10 лет. Кроме данного прогрессирующего младенческого типа заболевания, болезнь Александера также диагностируется у подростков и взрослых. Особенностью заболевания у пациентов старшего возраста является поражение ствола мозга и бульбарных структур.
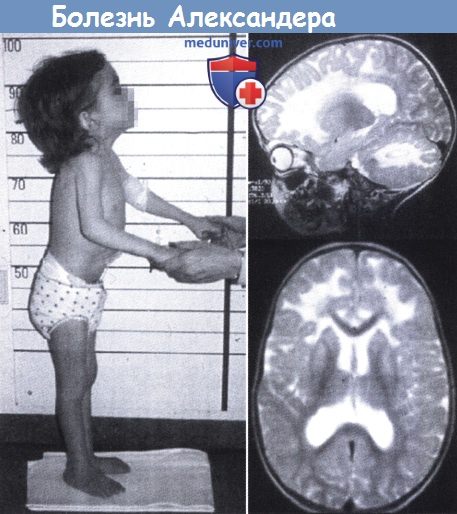
Болезнь Александера. Макроцефалия у четырехлетней девочки (слева).
На МРТ в сагиттальной проекции в Т2-режиме (справа) определяется повышение сигнала переднего белого вещества; заметно поражение мозжечка.
У пациентов старшего возраста чаще отмечается нормоцефалия. Иногда симптомы ошибочно диагностируются как новообразование ствола мозга. Описаны единичные семьи с несколькими больными, а также монозиготные близнецы (Meins et al., 2002). Заболевание было описано Mignot et al. (2004).
Диагностика классических случаев AXD основана на наличии макроцефалии, регрессии, припадков и бульбарных симптомов у пациентов старшего возраста.
На ЭЭГ можно выявить генерализованную медленную активность и некоторое количество очаговых разрядов. Наиболее ценную информацию можно получить по результатам КТ и МРТ. Описаны нейрорадиологические характеристики, которые практически все гда встречаются при AXD (Van der Knaap et al., 2001). Описанные признаки включают экстенсивное усиление сигнала мозгового белого вещества с преобладанием изменений в лобной области, перивентрикулярный ободок высокого сигнала в Т1-режиме и низкого сигнала в Т2-режиме, аномалии базальных ганглиев, таламуса и ствола мозга, а также усиление контрастирования отдельных структур серого и белого вещества.
При МР-спектроскопии выявляется выраженное накопление миоинозитола и снижение содержания α-нафтилуксусной кислоты, сопровождающееся накоплением лактата в пораженном белом веществе головного мозга и мозжечка. Уровень миоинозитола повышен в белом веществе, а также в сером веществе и базальных ганглиях.
Изменения отражают глиальную (астроцитарную) пролиферацию, а также активную демиелинизацию и нейроаксональную дегенерацию. Совсем недавно описано повышение GFAP в спинномозговой жидкости при всех трех подтипах AXD (Kyllerman et al., 2005). Диагноз подтверждается на основании молекулярно-генетической оценки гена GFAP.
В настоящее время проводится только симптоматическое лечение.