Классический вариант болезни Пелицеуса-Мерцбахера (PMD), сцепленный с Х-хромосомой, впервые был описан Пелицеусом в 1885 г. Мерцбахер в 1910 г. описал нейропатологию семьи, обследованной Пелицеусом, и пришел к выводу, что заболевание является результатом врожденной аплазии миелиновой оболочки. Значительно позже по результатам биохимического анализа белого вещества головного мозга было выявлено изменение, связанное с фетальным миелином, включающее снижение уровня протеолипидного белка (PLP). Seitelberger описал врожденный вариант заболевания (II тип). В соответствии с тяжестью заболевания выделяют также III тип.
Нейропатология классической формы болезни Пелицеуса-Мерцбахера (PMD) (I тип) характеризуется очаговой гипо- и дисмиелинизацией (так называемый тигроидный тип), в то время как при II типе отмечается практически полное отсутствие миелина (Seitelberger et al., 1995).
Делеции, дупликации и мутации гена PLP1 на хромосоме Xq22 являются причиной заболевания. Дупликации гена PLP1 могут быть выявлены у 20% пациентов с болезнью Пелицеуса-Мерцбахера (PMD), а точечные мутации — у 10-25% пациентов. Ген PLP1 дополнительно кодирует DM20 (белок ЦНС) (Sistermans et al., 1998).
По результатам недавнего исследования у пяти мальчиков с тяжелой врожденной формой PMD выявлено три и более копий протеолипидного белка PLP1 (Wolf et al., 2005). Отмечается также клиническая связь между легкими формами PMD и сцепленной с Х-хромосомой спастической параплегией 2 типа (SPG2), которая также является результатом мутаций гена PLP1.
Опубликованы клинические и радиологические критерии диагностики болезни Пелицеуса-Мерцбахера (PMD) (Boulloche и Aicardi, 1986). Критерии включают наличие у пациентов мужского пола отставания развитие моторики в возрасте до трех лет, чаще всего в возрасте нескольких месяцев, офтальмологические и неврологические изменения. Нистагм часто является первым выявляемым признаком. Гипотония, хореоатетоз, а затем и атаксия и спастичность развиваются постепенно. У большинства пациентов отмечается медленное улучшение до 10-12 лет. Большинство пациентов способны сидеть, некоторые могут ходить с поддержкой, но редко самостоятельно. Лишь некоторые дети способны говорить.
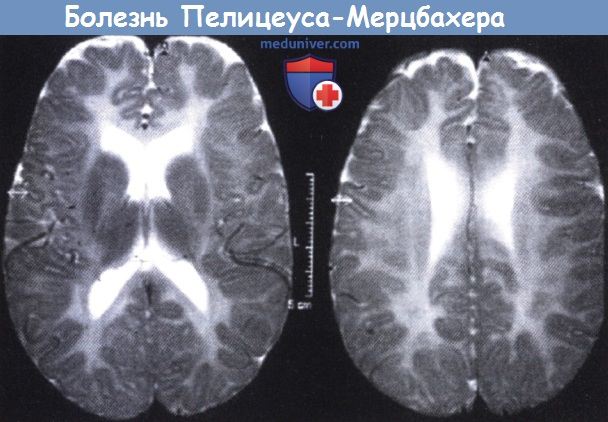
Болезнь Пелицеуса-Мерцбахера.
На МРТ в Т2-режиме определяется умеренно выраженное повышение сигнала перивентирулярного белого вещества.
Большинство пациентов достигает плато развития, а затем начинается медленная регрессия с дистонией, усиливающейся атаксией, спастичностью и формированием атрофии зрительного нерва.
При II типе заболевания (Haenggeli et al., 1989) отмечается более тяжелое течение с ограниченным развитием до начала ухудшения, хотя оно начинается практически в те же сроки, что и при классическом типе. У пяти пациентов в возрасте до трех месяцев развились тяжелые симптомы, включающие гипотонию, нистагм, стридор и припадки (Wolf et al., 2005). Два пациента умерли в возрасте 7 и 9 месяцев; трое страдали тяжелой эпилепсией.
Сочетание клинических, электрофизиологических и нейрорадиологических признаков является основой диагностики. Ранее формирование нарушения развития, нистагма, мышечной гипотонии и хореиформных движений являются характерными клиническими проявлениями. Нистагм может исчезать в возрасте до двух лет. Нарушение мозговой проводимости вызванных зрительных и/или слуховых потенциалов и диффузные области гиперинтенсивного сигнала на МРТ в Т2-режиме являются наиболее значимыми диагностическими признаками PMD.
Высокая интенсивность сигнала отмечается во всех немиелинизированных структурах белого вещества в Т2-режиме, а низкая интенсивность сигнала — в Т1-режиме. Тем не менее, параметры КТ могут быть обманчиво нормальными или же выявляется только некоторая атрофия. При МР-спектроскопии выявляются метаболические параметры белого вещества, сходные с параметрами серого вещества с повышением концентрации N-ацетиласпартата (NAA), глутамина, инозитола, креатина и фосфокреатина, в то время как концентрация холин-содержащих веществ снижается. Тем не менее, параметры МР-спектроскопии при PMD отличаются от показателей, выявляемых при других лейкодистрофиях (Hanefeld et al., 2005).
Фенотип болезни Пелицеуса-Мерцбахера (PMD) часто выявляется у пациентов, не имеющих мутаций гена PLP1, и в настоящее время классифицируется как PMD-подобное заболевание. Недавно было установлено, что мутации гена, кодирующего белок межклеточных щелевых контактов альфа-12 (коннексин 46.6), являются причиной PMD-подобного заболевания (Uhlenberg et al., 2004). В дополнение к PMD-подобной клинической картине у пациентов также отмечаются признаки периферической нейропатии и спорадические припадки. Данный вариант заболевания наследуется аутосомно-рецессивным путем. Иные атипичные формы данной клинической картины, вероятно, относятся к другим заболеваниям. Лечение только симптоматическое.