Нейродегенеративные заболевания - кратко с точки зрения внутренних болезней
Хотя рассеянный склероз (PC) является наиболее распространенной причиной инвалидизации молодых людей в Великобритании, сосудистые и нейродегенеративные заболевания приобретают все большее значение в более позднем возрасте. В основе нейродегенеративных заболеваний, распространенность которых повышается с возрастом, лежит патологический процесс, приводящий к специфической гибели нейронов и вызывающий неуклонное прогрессирование симптоматики. Причины этой группы заболеваний по-прежнему неизвестны, хотя вклад генетических факторов очень важен. Наиболее частыми из них являются болезнь Альцгеймера и БП.
а) Двигательные нарушения. Двигательные нарушения представлены широким спектром симптомов. Они могут быть наследственными или приобретенными; наибольшим образом они выражены при БП. Диагноз при большей части двигательных нарушений ставят на основании клинической картины, лабораторные и инструментальные исследования малоинформативны, за исключением случаев с известными генетическими мутациями.
1. Идиопатический паркинсонизм (болезнь Паркинсона). Паркинсонизм — это клинический синдром, характеризующийся прежде всего брадикинезией, повышением мышечного тонуса (ригидностью), тремором и утратой постуральных рефлексов. Обнаружено много причин синдрома паркинсонизма (табл. 54), но наиболее распространенной является БП. Ежегодная заболеваемость БП в Великобритании составляет около 18 на 100 000, а распространенность — примерно 180 на 100 000 населения.
Возраст является ключевым фактором, влияющим на заболеваемость и распространенность, причем последний показатель после 80 лет возрастает до 300—500 на 100 000 человек. Средний возраст начала заболевания составляет около 60 лет, и менее чем у 5% пациентов заболевание развивается до 40 лет.
Все больше признается роль генетических факторов, и были выявлены несколько отдельных генов, вызывающих паркинсонизм, хотя в целом они составляют очень небольшую долю случаев. Наличие родственника первой степени с БП повышает риск развития болезни в 2—3 раза. БП является прогрессирующей и неизлечимой, с различным прогнозом. Двигательные нарушения — наиболее распространенные проявления заболевания, но другие симптомы (такие как когнитивные нарушения, депрессия и беспокойство) становятся все более заметными по мере прогрессирования заболевания и значительно снижают качество жизни.
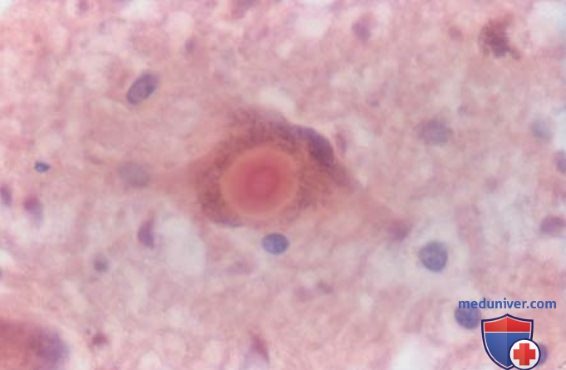
2. Патофизиология. Хотя в ряде случаев выявляются мутации в нескольких генах, у большинства пациентов причина заболевания остается неизвестной. Открытие того факта, что метилфенилтетрагидропиридин вызывает тяжелый паркинсонизм у молодых потребителей наркотиков, позволило предположить, что БП может быть связана с токсинами окружающей среды, но ни один из них не был достоверно идентифицирован. Патологическими признаками БП являются истощение пигментированных дофаминергических нейронов в черной субстанции и наличие α-синуклеина и других белковых включений в клетках черной субстанции (тельца Леви; рис. 1).
Рисунок 1. Болезнь Паркинсона. Черная субстанция пациента с болезнью Паркинсона при большом увеличении (х400): видно классическое тельце Леви (окраска гематоксилином и эозином).
Считается, что внешнесредовые или генетические факторы изменяют белок α-синуклеин, делая его токсичным и приводя к формированию телец Леви в клетках черной субстанции. Тельца Леви также обнаруживаются в базальных ганглиях, стволе головного мозга и коре, и их количество увеличивается по мере прогрессирования заболевания. БП обозначается как синуклеинопатия наряду с мультисистемной атрофией и деменцией с тельцами Леви. Утрата дофаминергической нейротрансмиссии вызывает многие клинические проявления.
3. Клиническая картина. Немоторные симптомы, в том числе снижение обоняния (гипосмия), беспокойство/депрессия, запор и нарушения поведения в фазе быстрого движения глаз, могут предшествовать развитию типичных моторных нарушений за долгие годы, но пациенты редко обращаются к врачу на этой стадии. Моторные симптомы почти всегда изначально асимметричны. Отличительной чертой является брадикинезия, приводящая к классическим симптомам: все более мелкий почерк («микрография»), трудности с завязыванием шнурков и застегиванием одежды, трудности с переворачиванием в постели.
Тремор является ранним признаком, но он может отсутствовать как минимум у 20% людей с БП. Обычно это односторонний тремор покоя, поражающий конечности, челюсть и подбородок, но не голову. У некоторых пациентов тремор остается доминирующим симптомом в течение многих лет. Ригидность вызывает тугоподвижность суставов и сгорбленность. Постуральные установочные рефлексы нарушаются на ранней стадии заболевания, но падения, как правило, не отмечаются вплоть до поздних стадий. По мере развития болезни речь становится все более тихой и нечеткой. При неврологическом обследовании обнаруживается ряд нарушений (табл. 55).
Изначально симптомы односторонние, но со временем развивается постепенное двустороннее поражение. На ранних стадиях заболевания когнитивная функция не страдает; при ее нарушении необходимо рассмотреть альтернативные диагнозы, такие как деменция с тельцами Леви.
- Немоторные симптомы. Хотя немоторные симптомы могут предшествовать появлению более типичных проявлений за долгие годы, у большинства пациентов они становятся все более распространенными и инвалидизирующими по мере прогрессирования БП. Нарушение когнитивных функций, включая деменцию, наиболее часто снижает качество жизни пациентов и лиц, осуществляющих уход за ними. Частота деменции составляет от 30 до 80% в зависимости от критериев диагноза и продолжительности наблюдения.
Другие беспокоящие пациента немоторные симптомы включают психоневрологические нарушения (тревога, депрессия, апатия, галлюциноз/психоз), расстройство сна и гиперсомния, повышенная утомляемость, боль, дисфункция сфинктеров и запоры, сексуальные проблемы (эректильная дисфункция, потеря либидо или гиперсексуальность), слюнотечение и снижение веса.
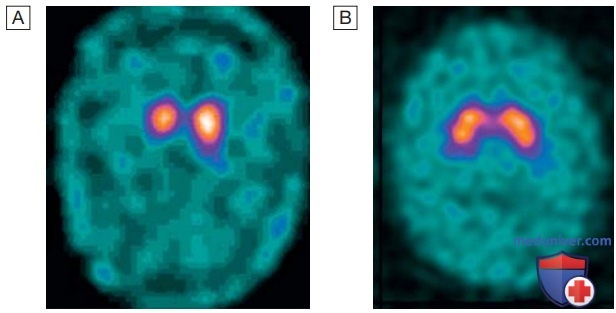
4. Лабораторные и инструментальные исследования. Диагноз ставится на основании клинической картины. Визуализация головного мозга (КТ или МРТ) обычно соответствует возрастной норме, и поэтому эти исследования редко бывают информативны, хотя и могут подтвердить подозреваемую сосудистую причину паркинсонизма. Функциональная дофаминергическая визуализация (однофотонная эмиссионная КТ или позитронно-эмиссионная томография) выявляет патологические изменения даже на ранних стадиях (рис. 2), но не позволяет дифференцировать различные формы дегенеративного паркинсонизма (см. табл. 54) и поэтому неспецифична для БП.
Рисунок 2. Результаты визуализации при болезни Паркинсона. А — однофотонная эмиссионная компьютерная томография при болезни Паркинсона, показывающая снижение активности дофамина в базальных ганглиях. В — норма
У более молодых пациентов может быть целесообразно применение специальных лабораторных и инструментальных исследований (например, исключение болезни Хантингтона или болезни Вильсона). Некоторые пациенты с отягощенным семейным анамнезом могут пожелать пройти генетическое тестирование, хотя роль генетического консультирования в настоящее время неясна.
5. Лечение:
5.1 Медикаментозная терапия. Медикаментозная терапия БП остается симптоматической и не приводит к излечению; отсутствуют данные о том, что какой-либо из имеющихся в настоящее время препаратов оказывает нейропротективное действие. Леводопа остается наиболее эффективным средством лечения, другие препараты включают агонисты дофамина, холино-блокаторы, ингибиторы моноаминоксидазы-В и катехол-О-метил-трансферазы и амантадин. Продолжаются споры о том, когда и какое лечение следует начинать. В целом большинство специалистов рекомендуют начинать терапию, когда симптомы нарушают повседневную жизнь, хотя некоторые предпочитают назначать препараты сразу после постановки диагноза.
Вопрос о том, с чего лучше всего начинать лечение — леводопой, агонистом дофамина или ингибитором моноаминоксидазы-В, — остается неясным, но большинство специалистов признают, что самым эффективным, переносимым и дешевым препаратом является леводопа. Многие моторные симптомы, такие как тремор, застывание, падения, наклон головы и патологическое сгибание, достаточно резистентны к лечению. Ряд немоторных симптомов (например, тревожность и депрессия) могут отвечать на медикаментозное или немедикаментозное лечение. В Великобритании ривастигмин лицензирован для использования при связанной с БП деменции, хотя его эффект остается умеренным. Многие другие немоторные симптомы также резистентны к проводимому лечению.
Препараты для БП не следует резко отменять, так как это может вызвать злокачественную гипертермию.
- Леводопа. Леводопа является предшественником дофамина. После приема внутрь более 90% декарбоксилируется на периферии (в желудочно-кишечном тракте и кровеносных сосудах) с образованием дофамина, и лишь небольшая часть достигает головного мозга. Это периферическое преобразование становится причиной высокой частоты нежелательных эффектов. Чтобы избежать этого, леводопу комбинируют с ингибитором дофа-декарбоксилазы; ингибитор не проникает через гематоэнцефалический барьер, что позволяет избежать нежелательного блокирования декарбоксилирования в головном мозге. Два ингибитора дофа-декарбоксилазы, карбидопа и бенсеразид, выпускаются в виде комбинированных препаратов с леводопой (препараты «Синемет» и «Мадопар» соответствен но).
Леводопа наиболее эффективна для уменьшения акинезии и ригидности мышц; действие на тремор часто менее выражено, и препарат не воздействует на многие моторные (поза, застывание) и немоторные симптомы. Если акинезия/ригидность не уменьшаются на фоне терапии леводопой (1000 мг/ сут), диагноз следует пересмотреть. Существуют лекарственные формы с контролируемым высвобождением леводопой, но их обычно лучше всего использовать на ночь, поскольку переменная биодоступность затрудняет их применение в течение дня. Леводопа + бенсеразид (Мадопар*) также выпускается в виде диспергируемой таблетки для ускорения наступления эффекта.
Нежелательные эффекты включают ортостатическую гипотензию, тошноту и рвоту, которые могут быть нивелированы домперидоном. Леводопа может усугублять или вызывать галлюцинации и, в редких случаях, патологическое поисковое поведение в отношении леводопы (синдром дофаминовой дисрегуляции), при котором пациент принимает чрезмерные дозы ЛД.
По мере прогрессирования БП реакция на леводопу у многих пациентов становится менее предсказуемой, что приводит к моторным флуктуациям. Это ухудшение состояния к концу действия дозы обусловлено прогрессирующей потерей емкости дофаминового хранилища из-за сокращения количества стриатонигральных нейронов. Индуцированные леводопой непроизвольные движения (дискинезия) могут возникать как феномен максимальной дозы или как двухфазный феномен (происходящий как на этапе наращивания, так и на этапе истощения эффекта дозы). Более сложные флуктуации представляют собой внезапные, непредсказуемые изменения реакции на лечение, при которых периоды паркинсонизма (фазы «выключения») чередуются с улучшением подвижности, но с дискинезиями (фазы «включения»).
Лечение моторных осложнений затруднено; феномен истощения эффекта дозы может отвечать на увеличение дозы или частоты приема леводопы или добавление ингибитора катехол-О-метил-трансферазы (см. ниже). Более сложные флуктуации могут улучшиться при добавлении агонистов дофамина (включая непрерывную инфузию апоморфина), использовании внутрикишечной формы леводопы с помощью чрескожной эндоскопической еюностомии или имплантации электродов для глубокой стимуляции головного мозга.
- Агонисты дофаминовых рецепторов. Первоначально они были разработаны в надежде отложить начало терапии леводопой и, следовательно, отсрочить моторные осложнения. Существуют несколько агонистов дофамина, которые могут назначаться внутрь, чрескожно или подкожно (табл. 56).
Агонисты из класса производных спорыньи больше не рекомендуются из-за редких, но серьезных фиброзирующих эффектов. За исключением апоморфина, все агонисты дофаминовых рецепторов значительно менее эффективны, чем леводопа, для уменьшения паркинсонизма, имеют больше нежелательных эффектов (тошнота, рвота, дезориентация и галлюцинации, расстройства привычек и влечений) и являются более дорогостоящими. Их рольвлечении БП (монотерапия или дополнительная терапия) остается неопределенной, и данные свидетельствуют о том, что их польза в качестве начальной монотерапии кратковременна.
- Ингибиторы моноаминоксидазы-В. Моноаминоксидаза типа В облегчает расщепление избытка дофамина в синапсе. При БП используются два ингибитора: селегилин и разагилин. Оба препарата умеренно эффективны, хотя обычно хорошо переносятся. Ни один из них не обладает нейропротек-тивным действием, несмотря на первоначально возлагаемые надежды.
- Ингибиторы катехол-О-метил-трансферазы. Катехол-О-метил-трансфераза (наряду с дофадекарбоксилазой) участвует в периферическом расщеплении леводопы. Существует два ингибитора — энтакапон и толкапон, который также ингибирует центральную катехол-О-метил-трансферазу. Энтакапон обладает умеренной активностью и наиболее эффективен при раннем истощении эффекта дозы. Он выпускается в виде отдельной таблетки, которую следует принимать вместе с каждой дозой леводопы / ингибитора дофа-декарбоксилазы, или в виде комбинированной таблетки с леводопой и ингибитором дофа-декарбоксилазы. Более сильнодействующий толкапон используется меньше из-за редкого, но серьезного побочного эффекта — гепатотоксичности.
- Амантадин. Этот препарат оказывает слабое, обычно кратковременное действие на брадикинезию и используется редко: только в том случае, если пациенты не могут переносить другие лекарственные средства. Он чаще применяется для лечения дискинезий, вызванных леводопой, хотя, опять же, его действие оказывается умеренным и кратковременным. К нежелательным эффектам относятся сетчатое ливедо, периферические отеки, делирий и другие антихолинергические эффекты.
- Холиноблокаторы. Это были основные препараты для лечения БП до появления леводопы. Их роль в настоящее время ограничена недостаточной эффективностью (помимо влияния на тремор в редких случаях) и нежелательными эффектами, включая сухость во рту, нечеткость зрения, запоры, задержку мочи, делирий и галлюциноз, а также отдаленные риски развития когнитивных нарушений. Выпускаются несколько холиноблокаторов, включая тригексифенидил (бензексол) и орфенадрин.
5.2 Хирургическое лечение. Деструктивные нейрохирургические операции широко использовались до появления леводопы. За последние 20 лет была разработана стереотаксическая хирургия, которая чаще всего включает глубокую стимуляцию головного мозга, а не деструктивный подход, характерный для предыдущих эпох. Были идентифицированы различные мишени, включая таламус (эффективно только при треморе), бледный шар и субталамическое ядро. Глубокая стимуляция головного мозга, как правило, показана у лиц с рефрактерным к медикаментозной терапии тремором или моторными флуктуациями, и тщательный отбор пациентов крайне важен для успеха лечения. Внутричерепная доставка фетальных трансплантатов или специфических факторов роста остается экспериментальным видом лечения.
5.3 Лечебная физкультура, трудотерапия и логопедическое лечение. На всех стадиях БП пациенты получают пользу от лечебной физкультуры, которая помогает уменьшить ригидность и корректирует неправильную позу. Специалисты по трудотерапии могут предоставить оборудование для преодоления функциональных ограничений, например перила для лестниц и туалета, а также оборудование для купания. Логопедическое лечение может помочь, когда дизартрия и дисфония мешают общению; также возможно проведение консультаций для пациентов с дисфагией. Как и при многих сложных неврологических расстройствах, пациентов с БП в идеале должна лечить многопрофильная команда, включая медсестер, прошедших специальное обучение в отношении БП.
б) Другие синдромы паркинсонизма. Цереброваскулярные заболевания и лекарственный паркинсонизм являются наиболее распространенными альтернативными причинами паркинсонизма (см. табл. 54). Несколько дегенеративных состояний также вызывают паркинсонизм и включают мультисистемную атрофию, прогрессирующий надъядерный паралич и кортикобазальную дегенерацию. Они обычно прогрессируют быстрее, чем БП, и резистентны к лечению леводопой. Эти заболевания определяются патогистологически, и идентификация в течение жизни затруднена. Есть и другие состояния, которые редко могут проявляться паркинсонизмом, в том числе болезни Хантингтона и Вильсона.
1. Мультисистемная атрофия. Мультисистемная атрофия характеризуется паркинсонизмом, вегетативной недостаточностью и мозжечковыми симптомами, причем преобладают либо паркинсонизм (МСА-П), либо мозжечковые симптомы (МСА-М). Мультисистемная атрофия встречается гораздо реже, чем БП, ее распространенность составляет около 4 на 100 000 человек. Хотя в начале заболевания различие между БП и МСА-П может быть затруднено, ранние падения, постуральная неустойчивость и отсутствие эффекта леводопы являются ее основными диагностическими критериями. Характерная морфологическая особенность заболевания — это содержащие α-синуклеин глиальные цитоплазматические включения в базальных ганглиях, мозжечке и моторной коре.
Лечение симптоматическое, и прогноз менее благоприятный, чем при БП, со средней выживаемостью от появления симптомов менее 10 лет и ранней инвалидизацией. Когнитивная функция обычно не страдает.
2. Прогрессирующий надъядерный паралич. Прогрессирующий надъядерный паралич характеризуется симметричным паркинсонизмом, когнитивными нарушениями, ранними падениями и бульбарными симптомами. Типичное нарушение движения глаз с медленными вертикальными саккадами, приводящими к ухудшению взора вверх и вниз, может развиваться несколько лет. Прогрессирующий надъядерный паралич имеет различные патологические особенности, связанные с аномальным накоплением тау- (т) белков и дегенерацией черной субстанции, субталамического ядра и среднего мозга. Следовательно, это таупатия, а не синуклеинопатия. Распространенность составляет около 5 случаев на 100 000 человек, при этом средняя выживаемость такая же, как при мультисистемной атрофии. Лечения не существует, и леводопа обычно не уменьшает выраженность паркинсонизма.
3. Кортикобазальная дегенерация. Кортикобазальная дегенерация встречается реже, чем мультисистемная атрофия или прогрессирующий надъядерный паралич, и ее клинические проявления могут быть различны, включая паркинсонизм, дистонию, миоклонус и феномен «чужой конечности», когда конечность (обычно верхняя) движется или мешает другой конечности без видимого сознательного контроля. Симптомы поражения коры головного мозга, включая деменцию и особенно апраксию, наблюдаются часто и в некоторых случаях могут быть единственными признаками. Ряд других заболеваний может проявляться кортикобазальным синдромом, включая другие деменции. Кортикобазальная дегенерация представляет собой таупатию с обширными отложениями в головном мозге; показатели выживаемости сходны с мультисистемной атрофией и прогрессирующим надъядерным параличом.
4. Болезнь Вильсона. Это аутосомно-рецессивное заболевание, возникающее в результате мутации в гене ATP7B, вызывающей нарушение метаболизма меди. Это излечимая причина различных двигательных нарушений, включая тремор, дистонию, паркинсонизм и атаксию; также возможно развитие психических расстройств. Болезнь Вильсона всегда следует исключать у пациентов в возрасте до 50 лет, страдающих какими-либо двигательными нарушениями.
5. Болезнь Хантингтона. Болезнь Хантингтона представляет собой аутосомно-доминантное заболевание, обычно встречающееся у взрослых, но иногда и у детей. Оно связано с экспансией тринуклеотидного CAG-повтора в гене на хромосоме 4. Болезнь часто характеризуется феноменом «предвосхищения», при котором она начинается во все более молодом возрасте по мере передачи из поколения в поколение из-за прогрессирующей экспансии повторов. Распространенность составляет около 4—8 случаев на 100 000 человек.
- Клиническая картина. Болезнь Хантингтона обычно сопровождается прогрессирующим расстройством поведения, патологическими движениями (обычно хореей) и когнитивными нарушениями, приводящими к деменции. Начало заболевания в возрасте до 18 лет встречается редко, но у пациентов может сначала появиться паркинсонизм, а не хорея (Вестфальский вариант болезни Хантингтона). У пациентов всегда отягощен семейный анамнез, хотя они могут его скрывать.
- Лабораторные и инструментальные исследования и лечение. Диагноз подтверждается с помощью генетического тестирования; возможно досимптомное тестирование для других членов семьи, но ему должно предшествовать соответствующее консультирование. Визуализация головного мозга может выявить атрофию хвостатого ядра, но этот метод диагностики ненадежен. Есть много заболеваний, напоминающих болезнь Хантингтона.
Лечение симптоматическое. Хорея может отвечать на терапию нейролептиками, такими как рисперидон, сульпирид или тетрабеназин. Депрессия и тревожность наблюдаются часто, и при них могут быть эффективны лекарственные препараты.
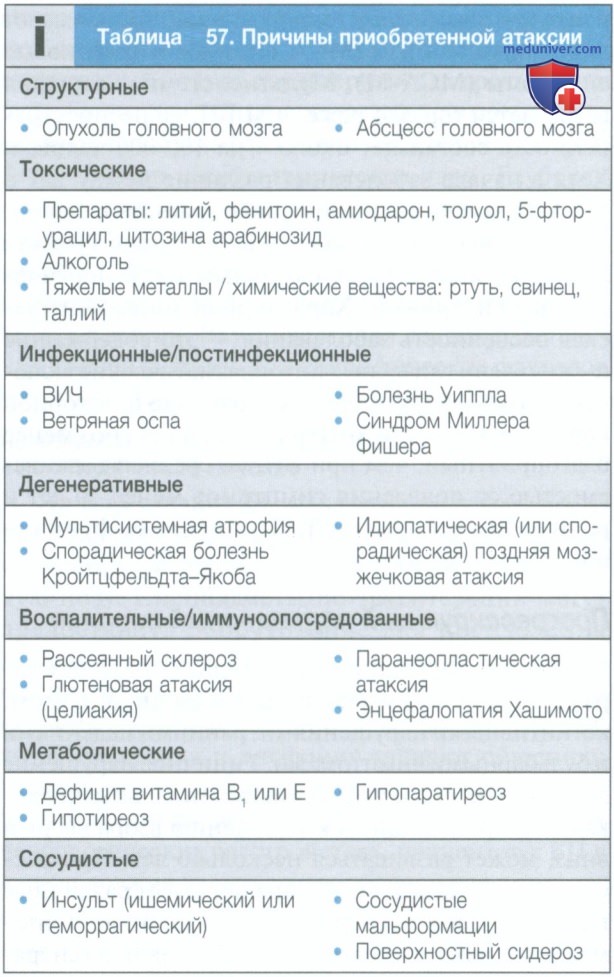
б) Атаксии. Атаксии представляют собой гетерогенную группу наследственных и приобретенных нарушений, проявляющихся изолированной атаксией либо сочетанием атаксии с другими неврологическими и иными симптомами. Дифференциальный диагноз нужно проводить с большим числом заболеваний (табл. 57 и 58), и постановка диагноза определяется возрастом дебюта болезни, ее эволюцией и клиническими проявлениями. У значительного числа пациентов, несмотря на проводимое обследование, приходится признать идиопатический характер нарушений.
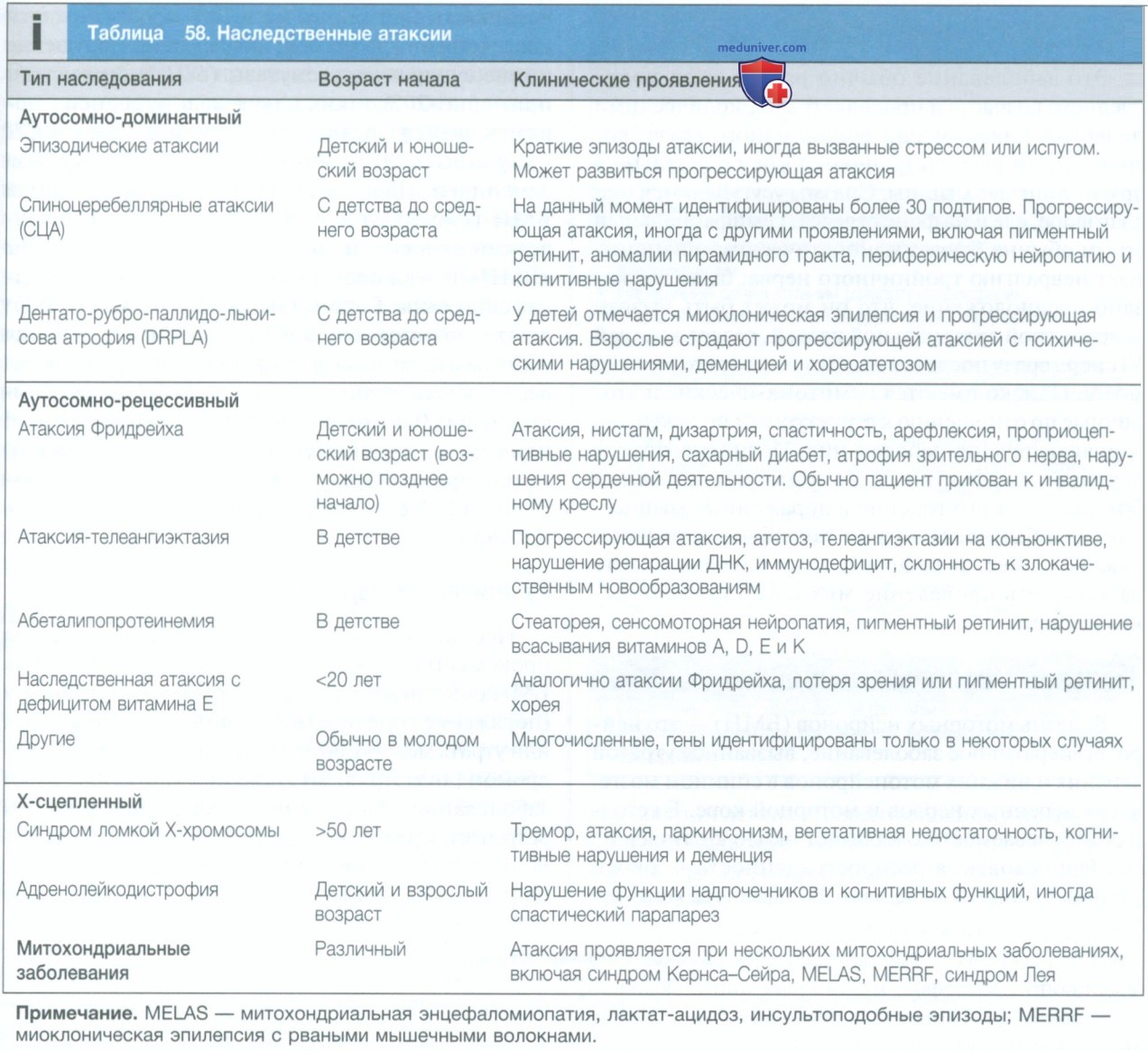
Наследственные атаксии представляют собой группу врожденных нарушений, при которых происходят дегенеративные изменения различной степени выраженности в мозжечке, стволе головного мозга, пирамидных путях, спиномозжечковых трактах, а также зрительных и периферических нервах, что определяет симптоматику. Заболевания могут дебютировать как в младенчестве, так и в зрелом возрасте, при этом тип наследования может быть рецессивным, сцепленным с полом и доминантным (см. табл. 58). Для некоторых вариантов были выявлены генетические нарушения, что позволило разработать диагностические тесты, но в настоящее время при многих наследственных атаксиях их проведение недоступно.
в) Тремор. Тремор — это признак многих расстройств, но самыми важными клиническими синдромами являются БП, эссенциальный тремор, лекарственный тремор (табл. 59) и функциональный (психогенный) тремор.
1. Эссенциальный тремор. Его распространенность составляет около 300 случаев на 100 000 человек, и он может наследоваться по аутосомно-доминантному типу, хотя генетические мутации, отвечающие за его развитие, до сих пор не идентифицированы. Эссенциальный тремор способен развиваться в любом возрасте в виде двустороннего тремора рук (8—10 Гц), редко отмечается в покое, обычно заметен при движении. В процесс могут вовлекаться голова и голосовые связки. Примерно у 50% пациентов тремор уменьшается после употребления небольшого количества алкоголя.
Специфических для данной патологии исследований нет, и эссенциальный тремор следует отличать от других синдромов тремора, включая дистонический тремор. Иногда эффективно применение бета-адреноблокаторов и примидона, а в тяжелых случаях — глубокая стимуляция таламуса.
г) Дистония. Дистония характеризуется фокальным повышением тонуса мышц конечностей или туловища. Она может быть характерной чертой ряда неврологических заболеваний (БП, болезнь Вильсона) или вторичным состоянием при повреждении головного мозга (травма, инсульт) или действии лекарственных препаратов (поздние синдромы). Дистония также возникает как первичное заболевание. Дистония, дебютировавшая в детстве, обычно генетически обусловлена и бывает генерализованной, но у взрослых ее начало чаще фокальное; в качестве примеров можно привести искривление шеи (кривошея), частое моргание глазами (блефароспазм) и тремор. Специфичные для выполнения различных задач симптомы (например, писчий спазм, спазм музыканта) часто имеют дистонию в своей основе. Лечение не разработано, но определенный эффект могут дать инъекции ботулинического токсина или глубокая стимуляция головного мозга.
д) Гемифациальный спазм. Это заболевание обычно развивается после среднего возраста и проявляется периодическими подергиваниями мышц вокруг одного глаза, которые затем распространяются ипсилатерально в другие лицевые мышцы. Спазмы усугубляются при разговоре, еде и на фоне стресса. Гемифациальный спазм обычно бывает идиопатическим и напоминает невралгию тройничного нерва; было высказано предположение, что он может быть вызван аберрантной артериальной петлей, раздражающей VII нерв сразу после его выхода из моста головного мозга. Однако имеются симптоматические и вторичные по отношению к структурным поражениям варианты течения заболевания. Медикаментозное лечение неэффективно, но помогают инъекции ботулинического токсина в пораженные мышцы, однако обычно их нужно повторять примерно каждые 3 мес. В рефрактерных случаях можно рассмотреть проведение микроваскулярной декомпрессии.
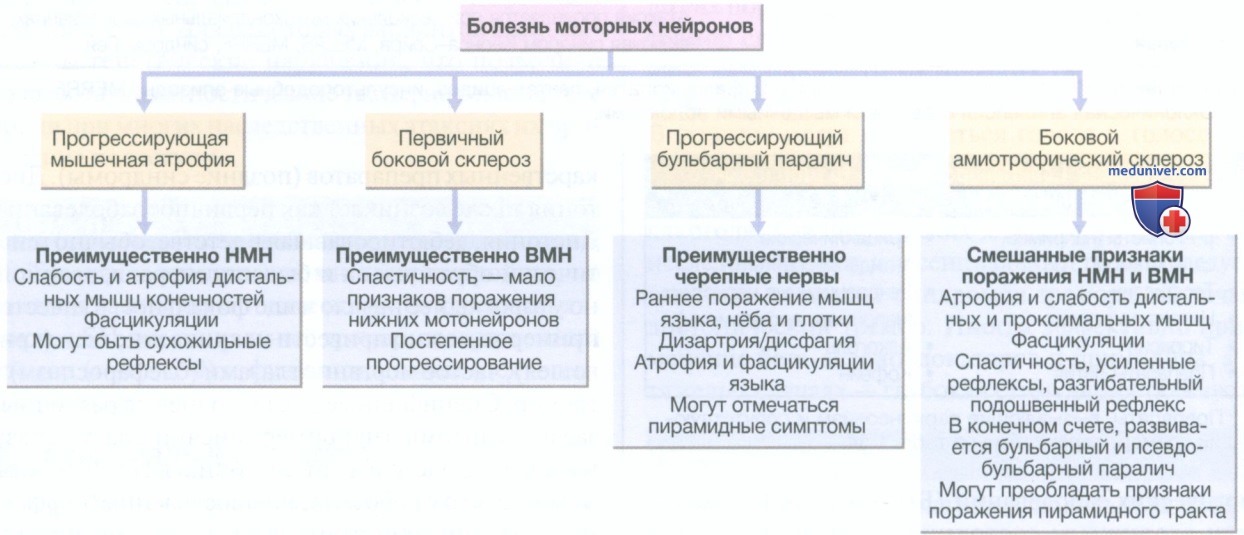
е) Болезнь моторных нейронов. Болезнь моторных нейронов (БМН) — это нейродегенеративное заболевание, вызванное утратой верхних и нижних мотонейронов в спинном мозге, ядрах черепных нервов и моторной коре. Ежегодная заболеваемость составляет около 2 случаев на 100 000 человек, а распространенность — около 7 случаев на 100 000 человек. Большинство случаев возникают спонтанно, но в 10% прослеживается наследственный характер заболевания. Нарушения в гене супероксиддисмутазы (SOD1) составляют примерно 20% таких случаев, и экспансия повторяющейся последовательности в гене C9orf72 на хромосоме 9 связана с БМН и лобно-височной деменцией. Наиболее распространенной формой БМН (рис. 3) является боковой амиотрофический склероз, и многие используют термины «БМН» и «боковой амиотрофический склероз» как синонимы.
Рисунок 3. Варианты поражения при болезни моторных нейронов. НМН — нижний мотонейрон; ВМН — верхний мотонейрон
Боковой амиотрофический склероз характеризуется сочетанием симптомов поражения верхних и нижних мотонейронов; существуют более редкие изолированные варианты БМН с поражением нижних (прогрессирующая мышечная атрофия) или верхних (прогрессирующий боковой склероз) мотонейронов. Средний возраст дебюта болезни — 65 лет, в 10% случаев заболевание манифестирует в возрасте до 45 лет.
1. Клиническая картина. Постановка диагноза бывает сложной и зачастую происходит несвоевременно. Клиническая картина БМН обычно дебютирует с фокальных нарушений (поражение конечности, например свисание стопы или утрата мелкой моторики) или бульбарным синдромом (дизартрия, затрудненное глотание); дебют заболевания с поражения дыхательной системы встречается редко, но дыхательная недостаточность II типа достаточно часто развивается в финале болезни. Сенсорные, вегетативные и зрительные симптомы обычно не возникают, а вот спазмы беспокоят часто (табл. 60). При обследовании выявляется комбинация признаков поражения нижних и верхних мотонейронов (например, оживленные рефлексы в атрофичных, фасцикулирующих мышцах) без сенсорных нарушений (см. рис. 3).
Когнитивные нарушения при БМН диагностируются плохо: почти у половины пациентов при стандартном тестировании выявляется преимущественно ухудшение исполнительных функций, и примерно у 10% развивается лобно-височная деменция. У 10% пациентов с лобно-височной деменцией боковой амиотрофический склероз развивается в течение нескольких лет после начала деменции. Даже на фоне лечения БМН неуклонно прогрессирует, но медиана выживаемости улучшается благодаря наблюдению специалистов, проводящих неинвазивную вентиляцию легких, осуществляющих питание пациента и назначающих при необходимости лекарственные препараты.
2. Лабораторные и инструментальные исследования. Клинические проявления, как правило, типичны, но необходимо исключить альтернативные заболевания. Очень важно исключение излечимых причин нарушений, таких как иммуноопосредованная мультифокальная моторная нейропатия с блоками проведения и шейная миелопатия. Анализы крови бывают, как правило, в норме, за исключением несколько повышенного уровня креатинфосфокиназы. Исследования скорости проведения импульса по сенсорным и моторным нервам не выявляет патологии, но может отмечаться снижение амплитуды моторных потенциалов действия из-за гибели аксонов.
ЭМГ обычно подтверждает типичные признаки широко распространившейся денервации и процесс реиннервации. Проведения анализа СМЖ обычно не требуется. Генетическое тестирование приобретает все большее значение, при этом обнаруживаются мутации в генах SOD1, FUS, TARDBP и C9orf72, которые могут помочь предсказать риск и фенотип заболевания у лиц с семейным анамнезом по БМН.
3. Лечение. Пациентов должна лечить многопрофильная команда, включающая специалистов по лечебной физкультуре, логопедов и специалистов по трудотерапии, диетологов, специалистов по искусственной вентиляции легких и парентеральному питанию, а также бригады паллиативной помощи с неврологической и респираторной поддержкой. Рилузол, антагонист высвобождения глутамата, лицензирован для лечения бокового амиотрофического склероза (в России не зарегистрирован), но обладает лишь умеренным эффектом, продлевая медиану выживаемости примерно на 2—3 мес.
Неинвазивная вентиляция легких значительно продлевает выживаемость и улучшает или поддерживает качество жизни у людей с боковым амиотрофическим склерозом. Выживаемость и ряд показателей качества жизни значительно улучшаются в подгруппе пациентов с исходно меньшей выраженностью бульбарного синдрома, по сравнению с пациентами с тяжелыми бульбарными нарушениями. Кормление через чрескожную гастростому может улучшить качество жизни и продлить выживаемость, даже если оно начинается на поздней стадии болезни. Быстрая доступность помощи со стороны врачей паллиативной помощи крайне важна при прогрессировании БМН до терминальных стадий.
ж) Спинальная мышечная атрофия. Это группа генетически детерминированных нарушений, поражающих нижние мотонейроны спинномозговых и черепных нервов и характеризующихся атрофией, фасцикуляциями и слабостью проксимальных и дистальных мышц. Поражение обычно симметрично, но иногда встречаются локализованные формы. За исключением детской формы, прогрессирование медленное и прогноз лучше, чем при БМН.