Советы по оценке КТ легких при врожденных заболеваниях
Существует множество врожденных заболеваний, которые могут приводить к различным патологическим изменениям органов грудной клетки. В некоторых случаях врожденные аномалии могут обусловливать появление выраженной клинической симптоматики, требующей коррекции. Аномалии могут также быть обнаружены случайно при лучевых исследованиях, выполненных по другим показаниям. Вне зависимости от проявлений лучевая диагностика играет значительную роль в идентификации и оценке врожденных аномалий органов грудной клетки, особенно легочной паренхимы и интерстиция. В некоторых случаях выявление преобладающей патологии, такой как легочный фиброз, кисты, бронхоэктазы, обызвествленные очаги, позволяет установить верный диагноз. В других случаях лучевая картина в совокупности с симптоматикой позволяет сформировать ограниченный дифференциально-диагностический ряд.
а) Лучевая диагностика. Первый метод диагностики, позволяющий обнаружить проявления врожденных заболеваний органов грудной клетки,-рентгенография. Тем не менее, КТ остается методом выбора при необходимости полной оценки патологических изменений, определения распространенности поражения и идентификации вероятных осложнений. Патологические процессы в легочной паренхиме и интерстиции могут приводить к различным изменениям на КТ, которые классифицируются по преобладающей патологии. Например, при семейном идиопатическом легочном фиброзе (СИЛФ) и синдроме Германски-Пудлака (СГП) основной находкой является фиброз легких. У пациентов с синдромом Берта-Хога-Дьюба преобладают воздушные легочные кисты. Микрокальцинаты являются основным проявлением альвеолярного микролитиаза, в то время как первичная цилиарная дискинезия (ПЦД) и муковисцидоз (МВ) характеризуются формированием бронхоэктазов.
б) Семейный идиопатический легочный фиброз (СИЛФ). СИЛФ-аутосомно-доминантное заболевание, характеризующееся клиническими проявлениями, сопоставимыми с идиопатическим легочным фиброзом (ИЛФ), а также изменениями на КТВР и гистологической картиной обычной интерстициальной пневмонии (ОИП). СИЛФ можно предположить при заболевании > 2 ближайших членов семьи (родителей, родных братьев и сестер, детей). Описаны множественные генетические мутации, связанные с данным заболеванием. Причинами СИЛФ также могут становиться внешние факторы (курение, воздействие металлической пыли, гастроэзофагеальный рефлюкс). Пациенты с СИЛФ обычно моложе пациентов с ИЛФ, тем не менее, исходы этих двух заболеваний аналогичны.
На КТВР/КТ СИЛФ проявляется классической картиной ОИП/ИЛФ, включая субплевральные ретикулярные изменения, «сотовое легкое», тракционные бронхо- и бронхиолоэктазы с апикально-базальным градиентом. Отличить СИЛФ и ИЛФ, опираясь лишь на КТ, невозможно. Для дифференциальной диагностики необходимо соотносить лучевую картину с персональным и семейным анамнезом.
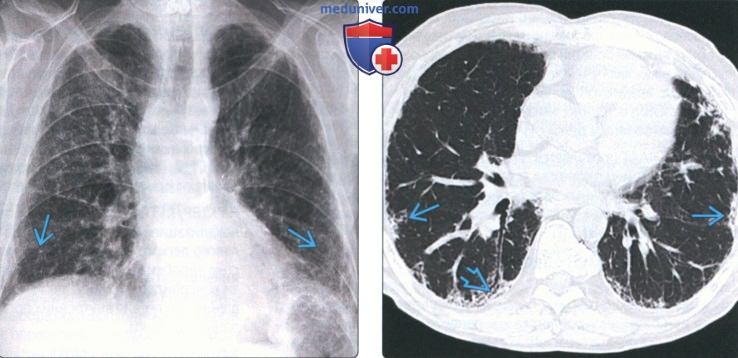
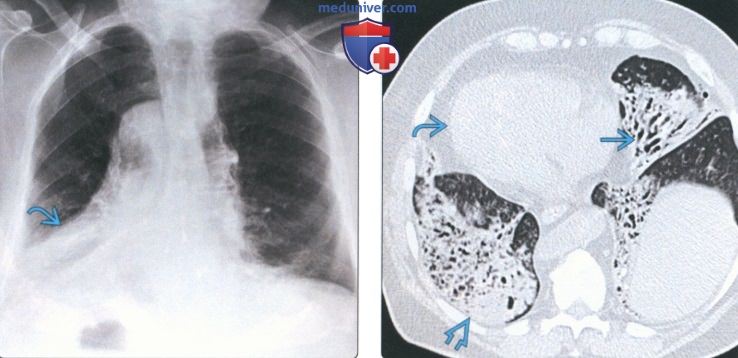
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у мужчины 47 лет с семейным идиопатическим легочным фиброзом определяются ретикулярные изменения, наиболее выраженные в базальных субплевральных отделах легких.
(Справа) На аксиальной КТ без КУ у этого же пациента определяется легочный фиброз наряду с тракционными бронхо- и бронхиолоэктазами с обеих сторон. Картина неотличима от спорадического идиопатического легочного фиброза. Дифференцировать данные заболевания только на основании лучевой картины возможно. Лучевые данные необходимо соотносить с персональным и семейным анамнезом.
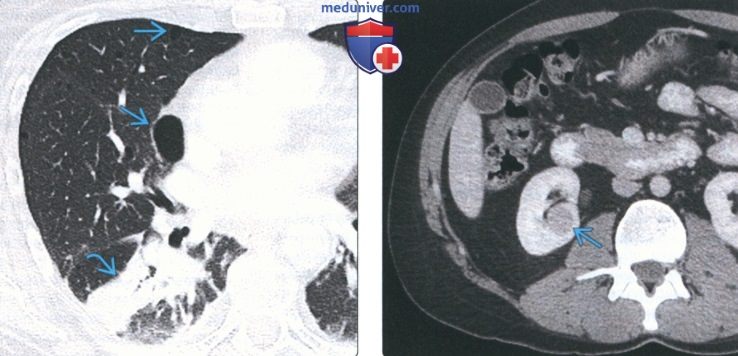
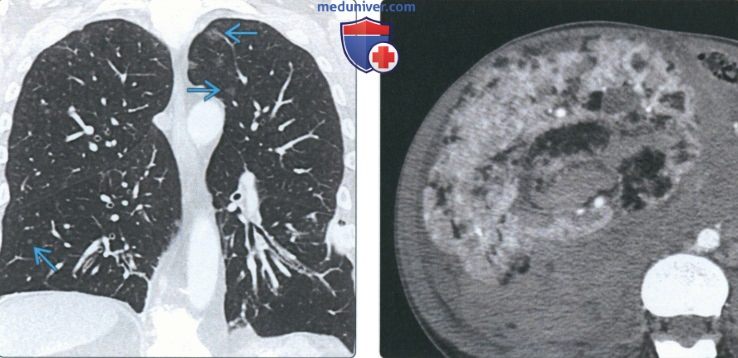
(Слева) На аксиальной КТ с КУ у женщины 41 года с синдромом Берта-Хога-Дьюба в правом легком определяются множественные тонкостенные кисты наряду с частичным ателектазом нижней доли.
(Справа) На аксиальной КТ с КУ органов брюшной полости у этой же пациентки в правой почке визуализируется объемное образование неоднородной структуры плотностью >50 единиц Хаунсфилда. У пациентов с синдромом Берта-Хога-Дьюба повышен риск возникновения злокачественных образований почек (почечноклеточного рака).
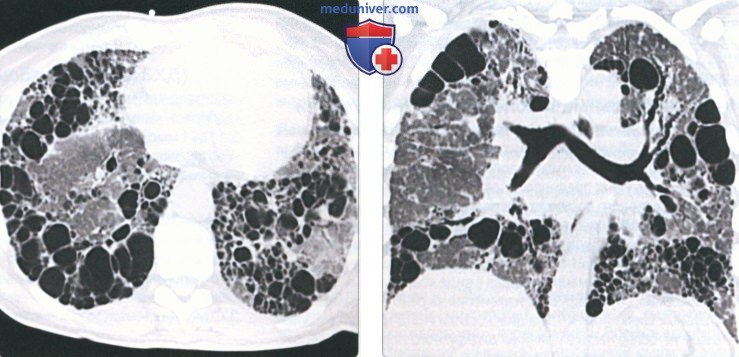
(Слева) На аксиальной КТ без КУ у пациента с синдромом Германски-Пудпака на поздней стадии в нижних долях обоих легких определяются множественные сгруппированные тонкостенные кисты на фоне «матового стекла».
(Справа) На корональной КТ без КУ у этот же пациента определяются множественные кисты, наиболее выраженные в нижних долях. К типичным проявлениям запущенных случаев заболевания относятся «сотовое легкое», субплевральные кисты, утолщение бронховаскулярных пучков, тракционные бронхо- или бронхиолоэктазы.
в) Синдром Берта-Хога-Дьюба (синдром БХД). Синдром БХД-аутосомно-доминантное заболевание, предрасполагающее к возникновению кист легких и почек, объемных новообразований почек, фиброфолликула из-за дефекта хромосомы 17р11.2 (FLCN) на участке, кодирующем фолликулин (белок-супрессоропухолей). Установлены специфические диагностические критерии синдрома БХД, подразделяющиеся на большие и малые.
Наиболее типичные патологические изменения в легких, обнаруживаемые на КТВР/КТ,- кисты с преимущественной локализацией в базальных отделах обоих легких (обнаруживаются в 89% случаев). Кисты обычно тонкостенные, имеют различные размеры, располагаются преимущественно в нижних долях легких вблизи плевры, в т.ч. междолевой, могут также прилежать к легочным сосудам. Синдром БХД может осложняться пневмотораксом.
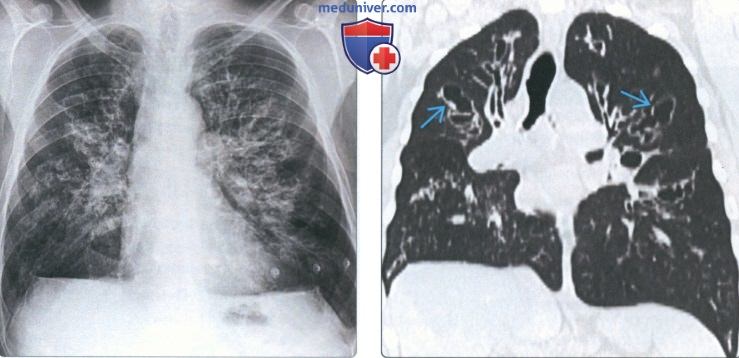
г) Синдром Германски-Пудлака (СГП). СГП-аутосомно-рецессивное генетическое заболевание, характеризующееся окулокутанным альбинизмом, дисфункцией тромбоцитов, скоплением цероид-липофусцина в лизосомах клеток различных органов и в ретикулоэндотелиальной системе. Идентифицированы десять различных типов СГП. Легочный фиброз, развивающийся у 50-70% пациентов, сильнее всего выражен при наиболее типичном СГП-1. В Америке СГП чаще всего наблюдается в Пуэрто-Рико, где заболеваемость достигает 1 на 1800 человек.
На КТВР/КТ специфические находки могут обнаруживаться на ранней и поздней стадии. К ранним проявлениям относятся утолщение междольковых перегородок, ретикулярные изменения, прикорневой фиброз, «матовое стекло». В запущенных случаях наблюдаются признаки «сотового легкого», субплевральные кисты, утолщение перибронховаскулярных тканей, тракционные бронхо- и бронхиолоэктазы. СГП может имитировать ИЛФ, хотя последний характеризуется преимущественным поражением нижних долей.
д) Туберозный склероз. Туберозный склероз (ТС) -мультисистемное аутосомно-доминант-ное генетическое заболевание, характеризующееся множественными гамартомами и злокачественными новообразованиями головного мозга, кожи, сетчатки, почек, сердца, легких. ТС-второй наиболее типичный факоматоз после нейрофиброматоза 1 типа (НФ1), возникающий из-за мутаций генов TSC1 и TSC2. Выделяют большие и малые диагностические критерии туберозного склероза, позволяющие установить достоверный, вероятный, предположительный диагноз ТС.
Наиболее типичные КТВР/КТ-признаки поражения легких-лимфангиолейомиоматоз (ЛАМ) и мультифокальная микронодулярная гиперплазия пневмоцитов (ММГП). ЛАМ проявляется так же, как и у пациентов без ТС,-тонкостенными кистами с диффузным распределением в обоих легких, количество и размеры которых увеличиваются со временем. ММГП проявляется множественными мелкими очагами в легких без обызвествлений в структуре, солидного характера или с плотностью «матового стекла». Вероятные осложнения: пневмо- и хилоторакс. Возможно также поражение других органов: сердца (очаговая жировая дегенерация миокарда, рабдомиомы), почек (ангиомиолипомы, кисты, почечноклеточный рак), костей (кистоподобные или остеобластические очаги, сколиоз).
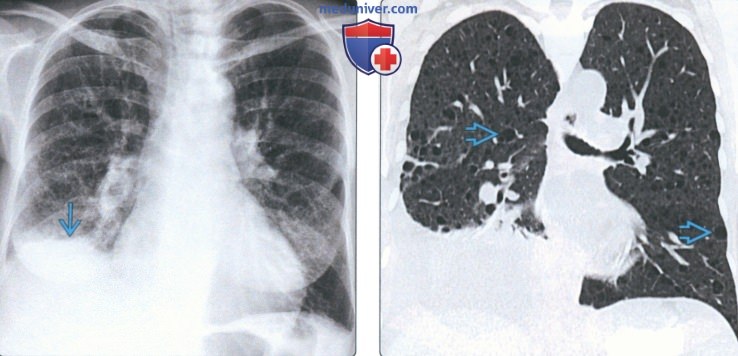
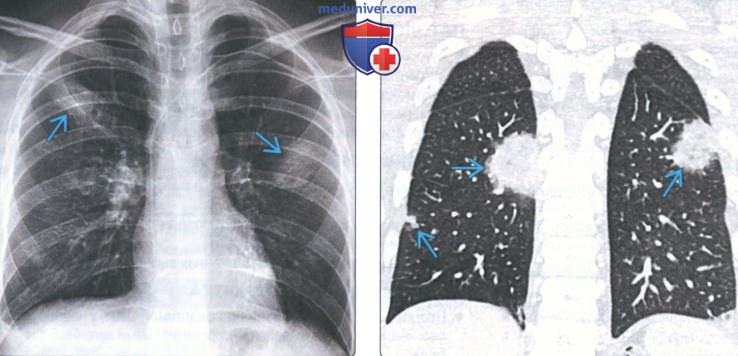
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у пациента с туберозным склерозом определяются ретикулярные изменения в легких, а также минимальный правосторонний гидроторакс.
(Справа) На корональной КТ с КУ у этого же пациента определяются множественные тонкостенные кисты с диффузным распределением в легких, а также минимальный-умеренный правосторонний гидроторакс. Наиболее типичные патологические изменения в легких, обнаруживаемые у пациентов с лимфангиолейомиоматозом, - тонкостенные кисты с диффузным распределением, размеры и количество которых увеличивается со временем.
(Слева) На корональной КТ с КУ у пациента с туберозным склерозом в обоих легких определяются множественные очаги с плотностью «матового стекла», сопоставимые с мультифокальной микронодулярной гиперплазией пневмоцитов. Очаги солидною характера и с плотностью «матового стекла» позволяют отличить туберозный склероз от лимфангиолейомиоматоза.
(Справа) На аксиальной КТ с КУ органов брюшной полости у этого же пациента определяются множественные ангиомиолипомы, практически полностью замещающие правую почку.
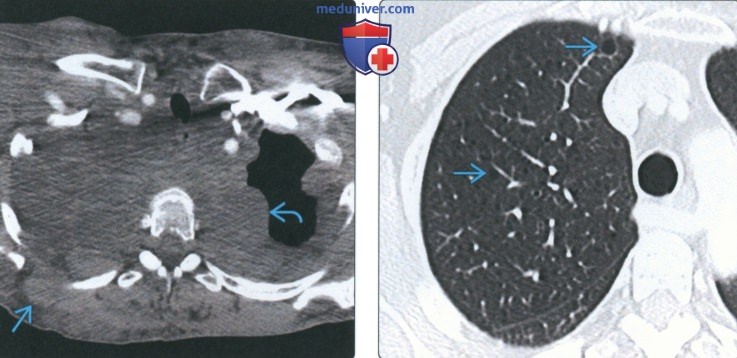
(Слева) На аксиальной КТ с КУ на уровне верхней трети грудной клетки у пациента с нейрофиброматозом 1 типа визуализируются множественные нейрофибромы грудной стенки и средостения, воздействующие на верхние доли обоих легких.
(Справа) На аксиальной КТ с КУ у пациента с нейрофиброматозом 1 типа определяются множественные тонкостенные кисты в правом легком. К дополнительным находкам относятся апикальные буллы и кисты, а также признаки легочного фиброза.
е) Нейрофиброматоз. НФ1 -аутосомно-доминантное генетическое заболевание, возникающее в результате мутации гена NF1 в 17 хромосоме, кодирующего синтез белка нейрофибромина. Это наиболее распространенный факоматоз, ассоциированный с системными проявлениями, который может приводить к поражению органов грудной полости. К «классическим» проявлениям НФ1 относятся доброкачественные опухоли оболочек нервов: нейрофиброма (фокальная форма) и плексиформная нейрофиброма (диффузная форма). Озлокачествление опухолей оболочек периферических нервов наблюдается у 2-16% пациентов с НФ1.
На КТВР/КТ у пациентов с нейрофиброматозом могут обнаруживаться тонкостенные апикальные буллы, кисты, проявления легочного фиброза (ретикулярные изменения, «сотовое легкое», тракционные бронхо- и бронхиолоэктазы). Редко встречающиеся внутрилегочные нейрофибромы выглядят как хорошо отграниченные очаги или объемные образования в легких. К сопутствующим находкам относятся внелегочные нейрофибромы и торакальное менингоцеле.
ж) Легочный альвеолярный микролитиаз. Легочный альвеолярный микролитиаз-редкое аутосомно-рецессивное заболевание, характеризующееся отложением в альвеолах узловых скоплений фосфата кальция (микролиты или калькосфериты), и обусловленное мутацией гена SLC34A2. У большинства пациентов на ранних стадиях заболевания симптоматика отсутствует, по мере прогрессирования в большинстве случаев развивается легочное сердце и дыхательная недостаточность.
Типичные проявления на КТВР/КТ: множественные плотные мелкие (< 1 мм) очаги в легких (микролиты), консолидация, «матовое стекло», утолщение междольковых перегородок и бронховаскулярных пучков, кисты. Множественные кальцинированные очаги в сочетании с утолщенными междольковыми перегородками могут создавать картину «сумасшедшей исчерченности».
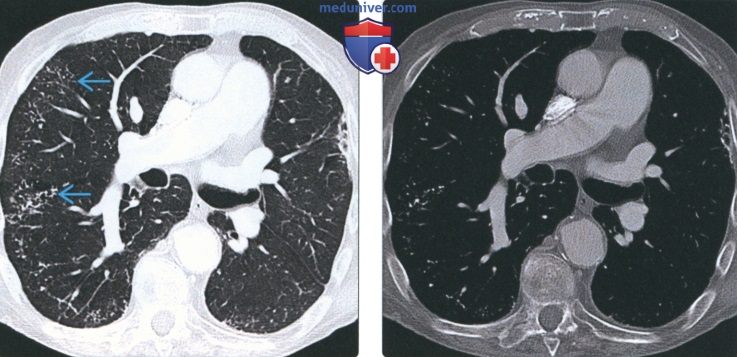
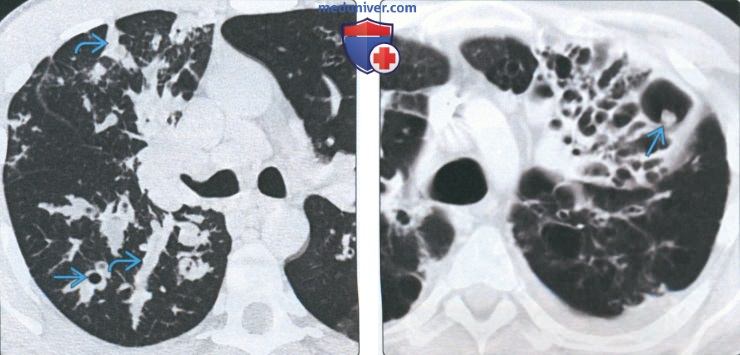
(Слева) На аксиальной КТ с КУ у пациента с легочным альвеолярным микролитиазом визуализируется множественные мелкие очаги в периферических отде пах обоих легких.
(Справа) На аксиальной КТ с КУ в костном окне у этого же пациента определяются обызвествления в большинстве данных очагов. Это нетипичное проявление легочного альвеолярного микролитиаза, имитирующее диффузную оссификацию легких.
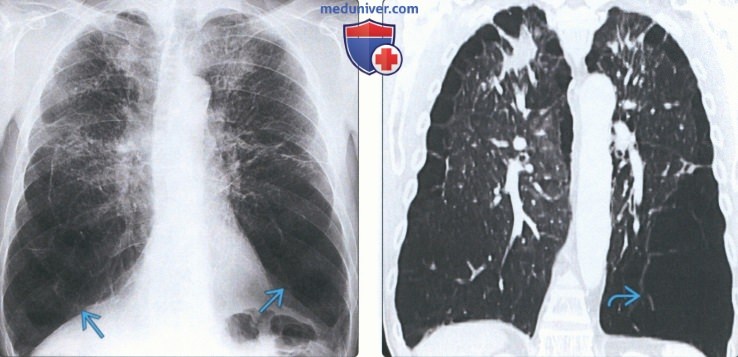
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у пациента с недостаточностью α-1 антитрипсина определяется вздутие легких с признаками эмфиземы, наиболее выраженными в нижних отделах.
(Справа) На корональной КТ с КУ у этого же пациента определяются признаки панлобулярной эмфиземы, наиболее выраженные в нижних долях. Снижена плотность легочной ткани, обеднен сосудистый легочный рисунок. Пациенты с недостаточностью α-1 антитрипсина предрасположены к раннему развитию хронической обструктивной болезни легких, цирроза и гепатоцеллюлярного рака.
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у пациента с синдромом Картатенера определяется декстрокардия и другие признаки транспозиции органов наряду с затемнениями в нижних отделах обоих легочных полей.
(Справа) На аксиальной КТ с КУ у этого же пациента в базальных отделах обоих легких на фоне декстрокардииопределяются множественные бронхоэктазы, затемнения по ходу бронховаскулярных пучков, участки консолидации. Картина сопоставима с первичной цилиарной дискинезией.
з) Недостаточность α-1 антитрипсина. Недостаточность α-1 антитрипсина (НААТ) — генетическое заболевание, возникающее в результате многочисленных нарушений гена SEPRINA1 (одноточечные мутации, инсерции, делеции). Люди с НААТ предрасположены к заболеваниям легких и печени, к раннему развитию обструктивной болезни легких (ХОБЛ), цирроза, злокачественных опухолей, таких как печеночноклеточный рак. Основное проявление НААТ-рано возникающая панлобулярная эмфизема. У 1-5% пациентов с ХОБЛ, вероятно, имеется НААТ. В отдельных случаях сообщается об эмфиземе у детей с НААТ.
На КТВР/КТ НААТ проявляется панлобулярной эмфиземой и равномерным снижением плотности легочной ткани с тотальным вовлечением вторичных легочных долек преимущественно в нижних долях. В пораженных участках чаще всего наблюдается обеднение сосудистого легочного рисунка.
и) Первичный иммунодефицит. Первичный иммунодефицит (ПИД) — группа наследуемых заболеваний, характеризующихся недоразвитием, отсутствием, дисфункцией одного или большего количества компонентов иммунной системы. Группа ПИД включает в себя > 200 различных заболеваний, симптомы и осложнения которых зависят от типа дефекта. Возможно преимущественное поражение придаточных пазух носа и среднего уха (синусит, средний отит) или нижних дыхательных путей и легких (пневмония, бронхит, бронхоэктазы). Могут также обнаруживаться признаки интерстициального заболевания легких (ИЗЛ).
КТВР/КТ позволяет обнаружить и дифференцировать множество патологических изменений в зависимости от этиологии иммунодефицита. Пневмония - наиболее типичное поражение легких, проявляющаяся сегментарными или долевыми участками консолидации, «матовым стеклом», «сумасшедшей исчерченностью». Поражение периферических дыхательных путей характеризуется «деревом в почках» и утолщением стенок бронхов. К вероятным осложнениям относятся пневматоцеле, абсцесс, легочное кровотечение.
к) Хроническая гранулематозная болезнь. Хроническая гранулематозная болезнь (ХГБ) — врожденное заболевание, приводящее к повышению чувствительности организма к инфекциям, обусловленное некоторыми бактериями и грибами. Специфический механизм ХГБ-дефект гена, кодирующего никотина-мидадениндинукпеотидфосфат-оксидазу, что приводит к снижению продукции кислородных радикалов, необходимых для внутриклеточного уничтожения микроорганизмов фагосомами. Х-сцепленная ХГБ наблюдается в 70% случаев, аутосомно-рецессивная ХГБ в 30%. Чаще всего ХГБ осложняется легочными инфекциями (пневмонией и абсцессом легкого).
На КТВР/КТ чаще всего обнаруживаются признаки легочной инфекции: участки консолидации, абсцесс, реактивная лимфаденопатия, внутрилегочные очаги, «дерево в почках», абсцесс средостения. У пациентов с рецидивирующими легочными инфекциями могут обнаруживаться рубцовые изменения в легких, тракционные бронхоэктазы и признаки эмфиземы.
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у юноши 19 лет с хронической гранулематозной болезнью определяются множественные участки консолидации в обоих легких. Присутствует так же минимальный правосторонний гидроторакс.
(Справа) На корональной КТ без КУ у этого же пациента определяются нодулярные и опухолеподобные участки консолидации с неровными контурами. Чаще всего у таких пациентов обнаруживаются признаки легочной инфекции: участки консолидации, внутрилегочные очаги, «дерево в почках», реактивная лимфаденопатия. Могут обнаруживаться осложнения: абсцесс легкого или средостения.
(Слева) На рентгенограмме органов грудной клетки в прямой проекции у пациента с муковисцидозом определяются центральные бронхоэктазы, наиболее выраженные в верхних и средних отделах легочных полей. Утолщены также стенки бронхов с обеих сторон.
(Справа) На корональной КТ без КУ у этого же пациента определяется выраженное расширение бронхов в верхних долях и менее выраженное в нижних долях. Такое распределение бронхоэктазов характерно для муковисцидоза.
(Слева) На аксиальной КТ без КУ у пациента с муковисцидозом в правом легком определяются множественные бронхоэктазы с внутрипросветными слизистыми пробками.
(Справа) На аксиальной КТ с КУ у пациента с муковисцидозом определяется выраженное расширение бронхов на отдельном участке в передних отделах верхней доли левого легкого. Внутрипросветный узел на задней стенке расширенного бронха представляет собой мицетому, обусловленную колонизацией бронха сапрофитными грибами.
л) Интерстициальные заболевания легких у детей. Интерстициальные заболевания легких у детей (ИЗЛд) - отдельная группа редких заболеваний легких, включающая в себе нарушения роста и развития, а также иммунологические нарушения, затрудняющие газообмен. ИЗЛд предполагается при обнаружении у младенца (< 2 лет) диффузного поражения легких в отсутствие других первичных заболеваний легких, при наличии как минимум трех из четырех перечисленных ниже критериев: (1) респираторная симптоматика (например, кашель); (2) признаки хронического заболевания легких (ретракция, деформация пальцев в виде «барабанных палочек); (3) гипоксемия; (4) диффузные изменения на рентгенограмме органов грудной клетки или КТ. Для диагностики ИЗЛд требуется исключить другие более частые причины диффузного поражения легких, такие как муковисцидоз, иммунодефицит, врожденные заболевания сердца, бронхолегочная дисплазия, легочная инфекция, первичная цилиарная дискинезия, рецидивирующая аспирация.
Изменения на КТВР/КТ могут в значительной степени варьировать и зависят от предшествующих заболеваний легких. Наиболее типичные нарушения: «матовое стекло», утолщение междольковых перегородок, кисты в легких, «мозаичная» картина, воздушные «ловушки».
м) Первичная цилиарная дискинезия (ПЦД). ПЦД-аутосомно-рецессивное генетическое заболевание, для которого характерно нарушение ультраструктуры ресничек, приводящее к мукоцилиарной дисфункции с вовлечением органа слуха, придаточных пазух носа, легких. Синдром Картагенера, проявляющийся триадой симптомов-транспозицией внутренних органов, синуситом и/или носовыми полипами, бронхоэктазами,- наблюдается в 50% случаев ПЦД. Функциональные нарушения дыхательных путей предрасполагают к рецидивирующим легочным инфекциям.
На КТВР/КТ ПЦД проявляется бронхоэктазами с преимущественной локализацией в средней доле правого легкого и язычковых сегментах левого легкого. Другие находки: утолщение стенок бронхов, слизистые «пробки», центрилобулярные очаги, «дерево в почках», участки консолидации. Могут также присутствовать сопутствующие нарушения положения внутренних органов, позволяющие предположить данный диагноз.
н) Муковисцидоз (МВ). МВ - аутосомно-рецессивное генетическое заболевание, характеризующееся нарушением транспорта хлоридов, в 25% всех случаев являющееся причиной возникновения бронхоэктазов у взрослых людей.
На КТВР/КТ бронхоэктазы являются преобладающей находкой с диффузным распределением и наиболее выраженным поражением верхних долей. Наиболее ранний признак МВ-утолщение стенок бронхов. Вздутие легких также относится к ранним проявлениям заболевания: в начальной стадии оно обратимо, но затем во всех случаях остается постоянным. Нужно отметить, что изменения на КТ больше соотносятся с клинической симптоматикой, чем с функцией легких.