Проявления иммунной недостаточности с вовлечением нескольких типов клеток могут варьировать от тяжелых до легких. Эти состояния сопровождаются тяжелой инфекцией, рецидивирующими инфекциями, редкими инфекциями или аутоиммунными нарушениями. Наиболее серьезное заболевание — тяжелый комбинированный иммунодефицит. К др. комбинированным иммунодефицитам относятся дефекты врожденного иммунитета и дефекты, ведущие к нарушению регуляции иммунитета.
Последняя категория обычно связана с глубоким иммунным нарушением. Комбинированные иммунодефициты отличаются предрасположенностью к вирусным инфекциям, а врожденные иммунодефициты — восприимчивостью к ряду бактериальных возбудителей.
Тяжелый комбинированный иммунодефицит возникает вследствие различных генетических мутаций, которые приводят к потере функции Т- и В-клеток. Пациенты с этой группой нарушений страдают самой тяжелой формой иммунодефицита.
а) Патогенез. Тяжелый комбинированный иммунодефицит возникает в результате мутаций в генах, ключевых для развития лимфоидных клеток (табл. 1 и рис. 1). У всех пациентов с тяжелым комбинированным иммунодефицитом очень маленькие тимусы, которые не содержат тимоцитов и не имеют кортикомедуллярных различий или телец Хассалла. Эпителий тимуса гистологически выглядит нормальным. Фолликулярная и паракортикальная области селезенки обеднены лимфоцитами. ЛУ, миндалины, аденоиды и пейеровские бляшки отсутствуют или крайне недоразвиты.
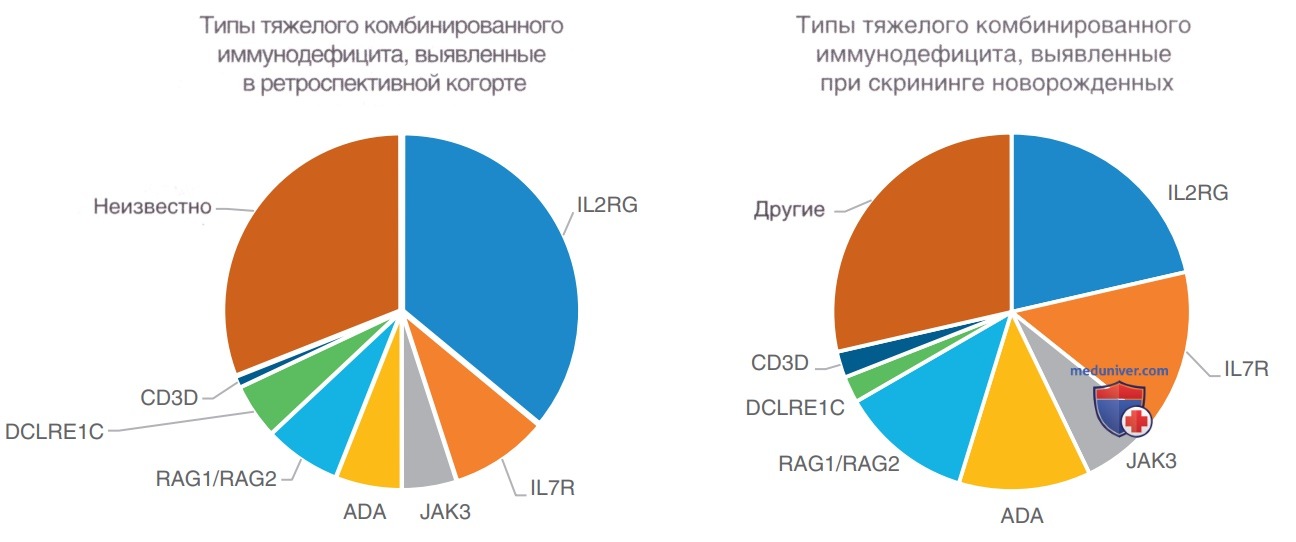
Рисунок 1. Относительные частоты встречаемости различных генетических типов тяжелого комбинированного иммунодефицита. ADA — аденозиндезаминаза; IL-7R — рецептор IL-7; JAK — янус-киназа; RAG — ген, активирующий рекомбиназу
б) Клинические проявления. Тяжелый комбинированный иммунодефицит включен в программу скрининга новорожденных во многих штатах. Младенцев выявляют до появления симптомов, что значительно улучшает выживаемость больных. Некоторые генетические типы тяжелого комбинированного иммунодефицита не обнаруживаются при скрининге новорожденных, а в нескольких штатах скрининг новорожденных еще не проводится.
Когда младенцы с тяжелым комбинированным иммунодефицитом не выявляются при скрининге новорожденных, у них чаще всего диагностируется инфекция. Наиболее частые проявления: диарея, пневмония, средний отит, сепсис и инфекции кожи. Инфицирование различными условно-патогенными микроорганизмами в результате прямого контакта или иммунизации может привести к летальному исходу.
Потенциальную угрозу представляют: Candida albicans, Pneumocystis jirovecii, вирус парагриппа 3, аденовирус, РСВ, ротавирусная вакцина, ЦМВ, ВЭБ, вирус ветряной оспы, вирус кори, комбинированная вакцина против кори, эпидемического паротита, краснухи и ветряной оспы (MMRV; англ. measles, mumps, rubella, varicella) или БЦЖ. У таких младенцев также отсутствует способность отторгать чужеродные ткани.
Они подвержены риску тяжелой или смертельной реакции «трансплантат против хозяина» из-за Т-лимфоцитов в необлученных продуктах крови или материнских иммунокомпетентных Т-клеток, которые проникали в/утробно трансплацентарно. Это злокачественное проявление характеризуется разрастанием аллогенных клеток, сыпью, гепатоспленомегалией и диареей. Третье проявление часто называют синдромом Оменна, при котором несколько клеток, генерируемых у младенца, пролиферируют и вызывают клиническую картину, аналогичную реакции «трансплантат против хозяина» (рис. 2).
Рисунок 2. Характерные клинические проявления синдрома Оменна у младенца. Обратить внимание на генерализованную эритродер-мию, шелушение кожи, алопецию и отеки.
Ключевая особенность тяжелого комбинированного иммунодефицита — низкое количество лимфоцитов, выявляемое почти у всех пациентов. Сочетание оппортунистических инфекций и неизменно низкого числа лимфоцитов — показание для обследования на наличие тяжелого комбинированного иммунодефицита.
Диагностическая стратегия как для младенцев с симптомами, так и для новорожденных, обнаруженных при скрининге, заключается в проведении проточной цитометрии для количественного определения Т-, В- и NK-клеток. Маркеры CD45RA и CD45RO информативны для ДД материнского приживления трансплантата и синдрома Оменна. Функцию Т-лимфоцитов определяют путем оценки пролиферативных ответов на стимуляцию.
Все генетические типы тяжелого комбинированного иммунодефицита ассоциируются с глубоким иммунодефицитом. У нескольких из них есть др. характерные или нетипичные особенности, которые важно распознать. Дефицит аденозиндезаминазы может быть связан с легочным альвеолярным протеинозом и хондрокостной дисплазией. Дефицит аденилаткиназы 2 (AK2) вызывает картину, называемую ретикулярным дисгенезом, когда нейтрофилы, миелоидные клетки и лимфоциты значимо снижены. Этому состоянию часто сопутствует глухота.
в) Лечение. Тяжелый комбинированный иммунодефицит — педиатрическая и иммунологическая неотложная ситуация. Если иммунологическое восстановление не достигается с помощью трансплантации гемопоэтических стволовых клеток или генной терапии, смерть наступает в течение первого года жизни и почти всегда до 2 лет. Трансплантация гемопоэтических стволовых клеток у младенца до инфицирования ассоциируется с 95% выживаемостью. Тяжелый комбинированный иммунодефицит, ассоциированный с дефицитом аденозиндезаминазы, и Х-сцепленный иммунодефицит успешно лечатся с помощью генной терапии.
Ранние испытания генной терапии были связаны с риском ЗНО, но этого не наблюдалось в испытаниях с новыми векторами. Тяжелый комбинированный иммунодефицит, ассоциированный с дефицитом аденозиндезаминазы, также можно лечить повторными инъекциями модифицированного полиэтиленгликоля бычьей аденозиндезаминазы. Успешное восстановление иммунитета более заметно при лечении с помощью стволовых клеток или генной терапии.
г) Генетика. Четыре наиболее распространенных типа тяжелого комбинированного иммунодефицита: Х-сцепленная форма, вызванная мутациями в CD132; АуР-дефицит RAG1 и RAG2, а также дефицит аденозиндезаминазы. Дополнительные формы перечислены в табл. 1. При Х-сцепленном тяжелом комбинированном иммунодефиците и дефиците аденозиндезаминазы применяется генная терапия, но генетическое консультирование — наиболее веская причина генетического секвенирования для определения дефекта гена.
Некоторые специфические генные дефекты связаны с повышенной чувствительностью к лучевой терапии и XT, и их раннее выявление может улучшить результат трансплантации.
Секвенирование выполняется путем создания панели генов-кандидатов на тяжелый комбинированный иммунодефицит. Разработаны определенные лабораторные критерии, которые позволяют прогнозировать специфические генные дефекты. Когда уровень Т- и В-клеток низкий, причина — кодирующий белок ген, участвующий в рекомбинации V(D)J. Точно так же определенные дефекты рецепторов цитокинов связаны со специфическими фенотипами лимфоцитов.
Гипоморфные мутации в генах, наиболее часто ассоциируемые с тяжелым комбинированным иммунодефицитом, могут приводить к появлению различных фенотипов. Это состояние часто называют «прободанный — дырявый — теряющий» тяжелый комбинированный иммунодефицит, имея в виду, что мутантный ген «прободан» для выработки некоторых лимфоцитов. «Прободанные» фенотипы варьируют от фенотипа синдрома Оменна к позднему иммунодефициту, гранулемам и аутоиммунным нарушениям.