Дефекты клеточного звена иммунитета, исторически относящиеся к дефектам Т-клеток, включают большую часть выраженных иммунодефицитов. Проявления — длительные вирусные инфекции, условно-патогенные грибковые или микобактериальные инфекции и предрасположенность к аутоиммунным заболеваниям.
Чтобы облегчить концептуальное представление этой большой и сложной категории, в этой статье на сайте описаны иммунодефициты, при которых дефект в первую очередь влияет на Т-клетки и те, у которых дефект изменяет функцию многих типов клеток. В отдельной статье на сайте описан тяжелый комбинированный иммунодефицит.
Эти заболевания далее рассматриваются с точки зрения клинического подхода с учетом наличия или отсутствия негематологических симптомов.
а) Синдром делеции хромосомы 22Q11.2. В США синдром делеции хромосомы 22q11.2 считается наиболее распространенным из Т-клеточных заболеваний, встречающимся у 1:3000 новорожденных. Делеция хромосомы 22q11.2 обуславливает нарушение развития 3-го и 4-го глоточных карманов во время раннего эмбриогенеза, что приводит к гипоплазии или аплазии тимуса и ПЩЖ.
Часто поражаются и др. структуры, формирующиеся в том же возрасте, в результате развиваются аномалии магистральных сосудов (правая дуга аорты), атрезии пищевода, раздвоение небного язычка, ВПС (дефекты конотрункальной перегородки, ДМЖП и ДМПП), короткий желобок верхней губы, гипертелоризм, антимонголоидный разрез глаз, гипоплазия нижней челюсти и повернутые кзади ушные раковины. Диагноз часто подтверждают во время гипокальциемических судорог в неонатальном периоде.
1. Генетика и патогенез. Делеции хромосомы 22q11.2 происходят часто: сложные повторяющиеся последовательности, фланкирующие область, — проблема для ДНК-полимеразы. Это заболевание наследуется по АуД-типу и встречается с сопоставимой частотой во всех группах населения. Гаплодостаточность по транскрипционному фактору ТВХ1 в удаленной области лежит в основе большинства фенотипов. Фенотип сильно варьирует.
У подгруппы пациентов обнаруживается фенотип, который также называют синдромом Ди Джорджи, велокардиофациальным синдромом или синдромом конотрункальной аномалии лица.
Вариабельная гипоплазия тимуса обнаруживается у 75% пациентов с делецией чаще, чем полная аплазия. Аплазия присутствует у <1% пациентов с синдромом делеции хромосомы 22q11.2. Менее половины пациентов с полной аплазией тимуса гомозиготны по хромосоме 22q11.2. Примерно 15% рождаются от матерей, страдающих СД. Еще 15% младенцев не имеют установленных факторов риска.
Примерно 1/3 младенцев с полным синдромом Ди Джорджи имеет CHARGE-синдром [англ/ аббревиатура CHARGE используется для обозначения новорожденных детей с врожденными признаками колобомы глаза (С), пороками сердца (Н), атрезией носовых хоан (А), задержкой роста и развития (R), гипоплазией половых органов (G) и аномалией уха (Е), включая глухоту]. Мутации в гене геликазного хромодомена ДНК-связывающего белка 7 (CHD7) на хромосоме 8ql2.2 обнаруживаются примерно у 60-65% людей с CHARGE-синдромом; у меньшинства обнаруживаются мутации в SEMA3E.
Абсолютное количество лимфоцитов умеренно снижено. Количество CD3 Т-клеток варьирует в зависимости от степени гипоплазии тимуса. Ответы лимфоцитов на стимуляцию митогена отсутствуют, снижены или нормальны, в зависимости от степени тимической недостаточности. Уровни иммуноглобулинов часто в норме, но отмечена повышенная частота дефицита IgA, низкие уровни IgM, а у некоторых пациентов развивается прогрессирующая гипогаммаглобулинемия.
2. Клинические проявления. Дети с частичной гипоплазией тимуса могут легко переносить инфекции, патология не влияет на их рост. Пациенты с аплазией тимуса напоминают пациентов с тяжелым комбинированным иммунодефицитом по своей восприимчивости к инфекциям, вызванным низко-или условно-патогенными микроорганизмами, включая грибы, вирусы и Pneumocystis jirovecii, а также к реакции «трансплантат против хозяина» в результате переливания необлученной крови.
У пациентов с полным синдромом Ди Джорджи может развиться атипичный фенотип, при котором в крови появляются популяции олигоклональных Т-клеток, ассоциированных с появлением сыпи и лимфаденопатии. Эти атипичные пациенты выглядят фенотипически подобными пациентам с синдромом Оменна или с приживлением материнских Т-лимфоцитов.
Важно своевременно диагностировать у младенца аплазию тимуса, поскольку без лечения это заболевание приводит к летальному исходу. Подсчет Т-клеток следует проводить у всех младенцев, рожденных с первичным гипопаратиреозом, CHARGE-синдромом и конотрункальными сердечными аномалиями с синдромальными особенностями. Некоторых младенцев выявляют при скрининге новорожденных на тяжелый комбинированный иммунодефицит, и при подозрении на делецию 22q11.2 необходимо определить уровень кальция во время определения Т-клеток.
Три проявления с самой высокой заболеваемостью в раннем младенчестве — глубокий иммунодефицит, тяжелая сердечная аномалия и судороги вследствие гипокальциемии. Необходимо сосредоточить внимание на этих проблемах еще до подтверждения диагноза. У больных могут развиться аутоиммунные цитопении, ЮНА, атопия и ЗНО (лимфомы).
3. Лечение. Иммунодефицит при аплазии тимуса корректируется трансплантацией культивированных неродственных клеток тимуса. Некоторым младенцам с аплазией тимуса сделана трансплантация необлученного и нефракционированного костного мозга или стволовых клеток периферической крови от идентичных по HLA кровных доноров (родных братьев или сестер) с последующим улучшением иммунной функции посредством адаптивно перенесенных Т-клеток.
Младенцы и дети с низким уровнем Т-лимфоцитов (но не в такой степени низким, чтобы рассматривать возможность трансплантации) должны находиться под постоянным наблюдением на предмет развития дефицита иммуноглобулинов. Причины инфекционных заболеваний у этих пациентов многофакторны. Их анатомические особенности не благоприятствуют дренажу секрета; они имеют более высокий риск развития атопии, что может осложнить течение инфекционного процесса; защитные реакции организма увеличивают риск персистенции инфекций.
Терапевтические рекомендации варьируют от гигиены рук, назначения пробиотиков, профилактической АБТ, контроля рисков заболевания до заместительной терапии иммуноглобулинами больных с выявленным дефектом гуморального иммунитета.
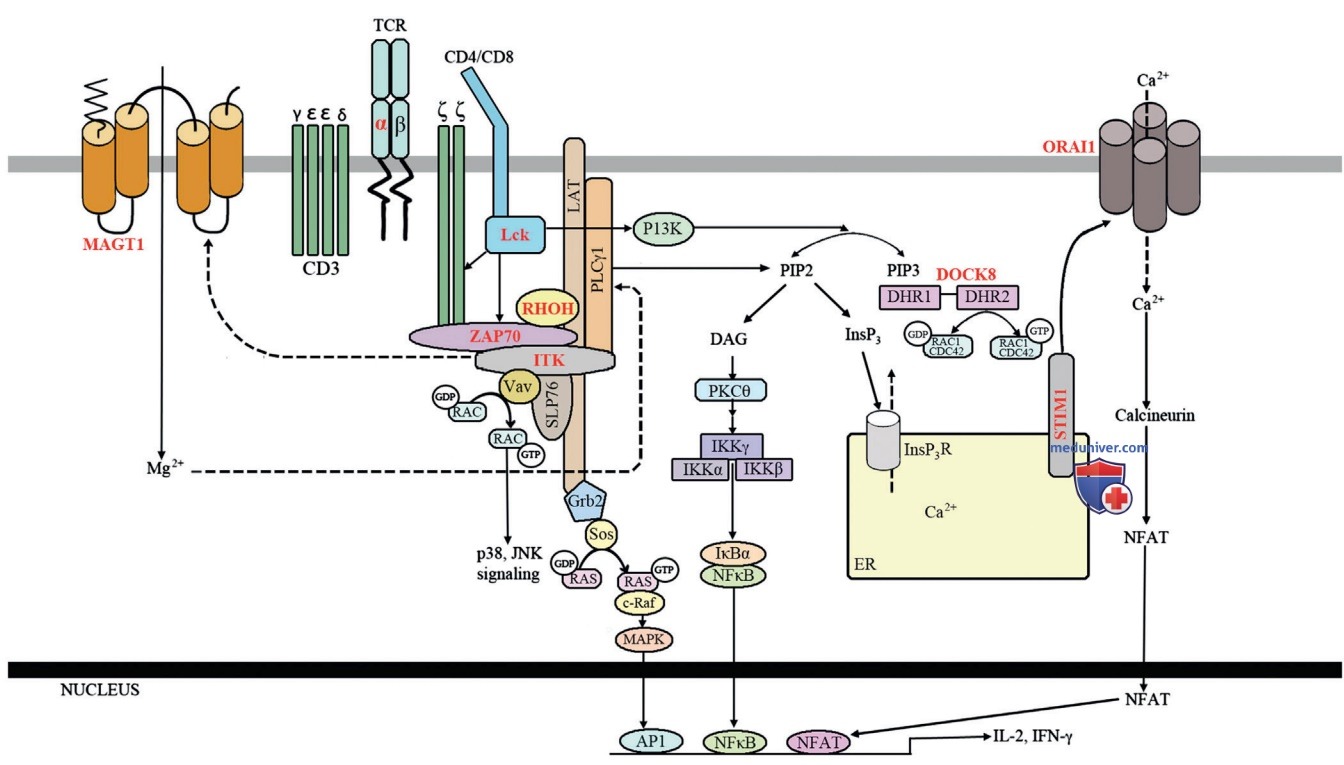
б) Нарушения активации Т-клеток. Нарушения активации Т-клеток характеризуются наличием нормального или повышенного уровня количества Т-клеток крови, которые кажутся фенотипически нормальными, но не могут нормально пролиферировать или продуцировать цитокины в ответ на стимуляцию митогенами, АГн или др. сигналами, доставляемыми в TCR-рецептор, из-за нарушения передачи сигнала от TCR-рецептора к в/клеточным метаболическим путям (рис. ниже).
Схематическое изображение передачи сигналов через комплекс Т-клеточный рецептор-CDЗ. Молекулы, мутации которых были связаны с частичным дефектом развития Т-клеток и нарушением Т-клеточной функции, выделены красным и жирным шрифтом. АР1 — белок-активатор 1; DHR — область гомологии DOCK;Grb2 — белок 2, связанный с рецептором фактора роста; IKK — 1кВ-киназа; JNK— N-концевая киназа c-Jun; MAPK — митогенактивированная протеинкиназа; NFAT — ядерный фактор активированных Т-клеток; NF-кВ — ядерный фактор кВ; PI3K — фосфоинозитид-3 киназа; PIP3 — фосфатидилинозитол(3,4,5)трифосфат.
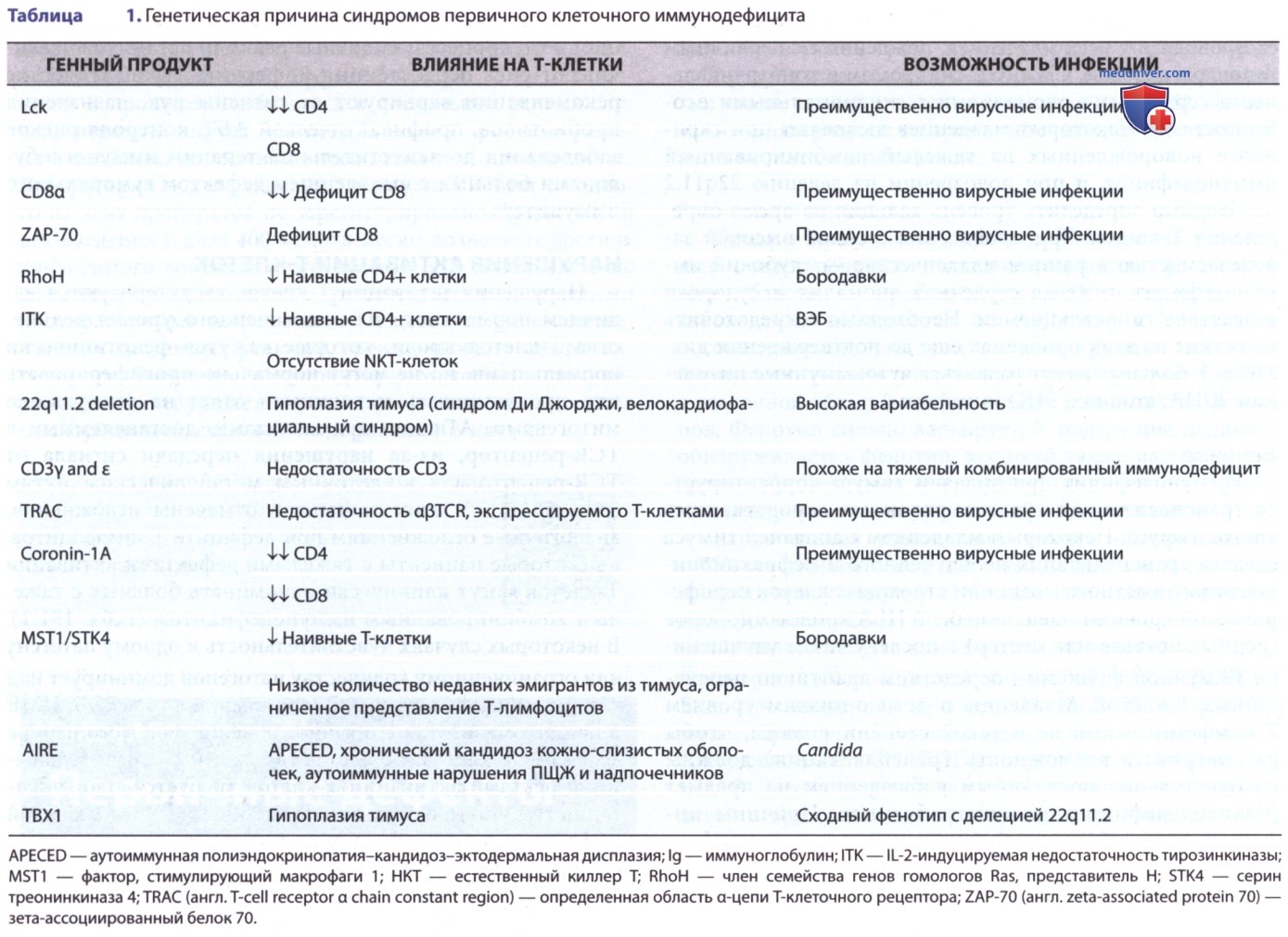
У этих пациентов отмечены осложнения, аналогичные осложнениям при дефиците Т-лимфоцитов, а некоторые пациенты с тяжелыми дефектами активации Т-клеток могут клинически напоминать больных с тяжелым комбинированным иммунодефицитом (табл. 1). В некоторых случаях чувствительность к одному патогену или ограниченному количеству патогенов доминирует над клиническим фенотипом. Восприимчивость к ВЭБ, ЦМВ и папилломавирусу — обычное явление для этого набора дефектов Т-клеток.
Большинству людей со значительными дефектами активации Т-клеток требуется трансплантация гемопоэтических стволовых клеток. Хотя с каждой инфекцией можно справиться в раннем возрасте, долгосрочный прогноз при большинстве этих заболеваний неблагоприятен.
в) Хронический кожно-слизистый кандидоз. Хронический кожно-слизистый кандидоз — синдром, характеризующийся нарушенной иммунной реакцией на грибы рода Кандида (Candida spp.). Пациенты-носители некоторых из известных генных мутаций в дополнение к хроническому кожно-слизистому кандидозу имеют аутоиммунный полиэндокринный синдром 1-го типа (аутоиммунная полиэндокринопатия-кандидоз-эктодермальная дистрофия или APECED-синдром).
Один из др. генетических типов хронического кожно-слизистого кандидоза ассоциируется с аутоиммунными нарушениями и предрасположенностью к инфекциям (мутации, связанные с усилением функции STAT1). Однако большинство определенных генетических типов хронического кожно-слизистогО кандидоза имеют индивидуальную восприимчивость к грибам рода Candida.
Эти типы связаны с дефектами пути дифференцировки Th17 клеток. АуР-дефицит в цепи рецептора IL-17RA и АуД-дефицит цитокина IL-17F ассоциируются с предрасположенностью к заболеваниям, вызванных Candida spp. Иные иммунодефициты, при которых кандидозы возникают в ассоциации с др. инфекциями, также влияют на клетки Th17. Др. генетический тип хронического кожно-слизистого кандидоза, вызванный мутациями в CARD9, ассоциируется с высоким риском развития не только кандидоза (Candida spp.), но и др. грибковых инфекций.
Лежащие в основе заболевания генные мутации различны, но клиническая картина хронического кожно-слизистого кандидоза обычно схожа. Симптомы могут появиться в течение 1 мес жизни или позднее (на протяжении 2-го десятилетия). Заболевание характеризуется хроническими, тяжелыми инфекциями кожи и слизистых оболочек, вызванными грибами рода Candida. У пациентов редко развивается системный кандидоз, за исключением случаев, указанных ниже. Местная противогрибковая, может обеспечить незначительное улучшение на ранней стадии заболевания, однако требуются системные курсы азолов.
Противогрибковая резистентность часто развивается в более позднем возрасте. Инфекция временно поддается лечению, санация возбудителя кратковременная с последующими рецидивами. Пациенты с мутациями гена CARD9 имеют более серьезную восприимчивость к грибкам, чем пациенты с хроническим кожно-слизистым кандидозом. У двух описанных пациентов с мутациями CARD9 отмечен грибковый сепсис в дополнение к хроническому кожно-слизистому кандидозу; также наблюдались глубокие поражения тканей дерматофитами.
г) Аутоиммунная полиэндокринопатия-кандида-эктодермальная дисплазия. Пациенты с этим синдромом страдают хроническим кожно-слизистым кандидозом и аутоиммунной полиэндокринопатией, в зрелом возрасте у них развивается гипопаратиреоз и болезнь Аддисона. Дополнительные проявления — мужской и женский гипогонадизм, хронический активный гепатит, алопеция, витилиго, пернициозная анемия, гипоплазия эмали, СД-1, аспления, мальабсорбция, интерстициальный нефрит, гипотиреоз, гипопитуитаризм и синдром Шегрена. APECED-синдром вызывается мутацией в гене аутоиммунного регулятора (AIRE) (табл. 1).
Продукт гена AIRE экспрессируется на высоких уровнях в очищенных стромальных клетках мозгового вещества тимуса человека. Он регулирует экспрессию на клеточной поверхности тканеспецифичных белков инсулина и тиреоглобулина. Экспрессия этих собственных белков обуславливает отрицательный отбор аутореактивных Т-клеток во время их развития. Отсутствие "-"-отбора приводит к органоспецифической аутоиммунной деструкции.