Нарушения выработки АТл наиболее распространены среди первичных иммунодефицитных заболеваний. Селективное отсутствие IgA — одна из наиболее распространенных патологий, ее частота среди представителей разных рас и этнических групп колеблется от 1/333 до 1/18 000 человек.
Пациентов с дефицитом АТл выявляют, потому что они страдают от рецидивирующих инфекций ВДП и НДП. Некоторые люди с избирательным дефицитом IgA или младенцы с транзиторной гипогаммаглобулинемией могут вообще не страдать от инфекций. Это заболевания с комплексным и полигенным наследованием, как и синдромы общего вариабельного иммунодефицита.
Дефекты генов ответственных за развитие многих заболеваний, связанных с первичными нарушениями продукции АТл, были идентифицированы (табл. 1) и локализованы (рис. 1). Иногда дефект находится не в самой В-клетке, а в Т-клетках, которые необходимы для полноценного функционирования В-клеток. Некоторые болезни вызваны неизвестными факторами или вторичны по отношению к основному заболеванию или его лечению (табл. 2).
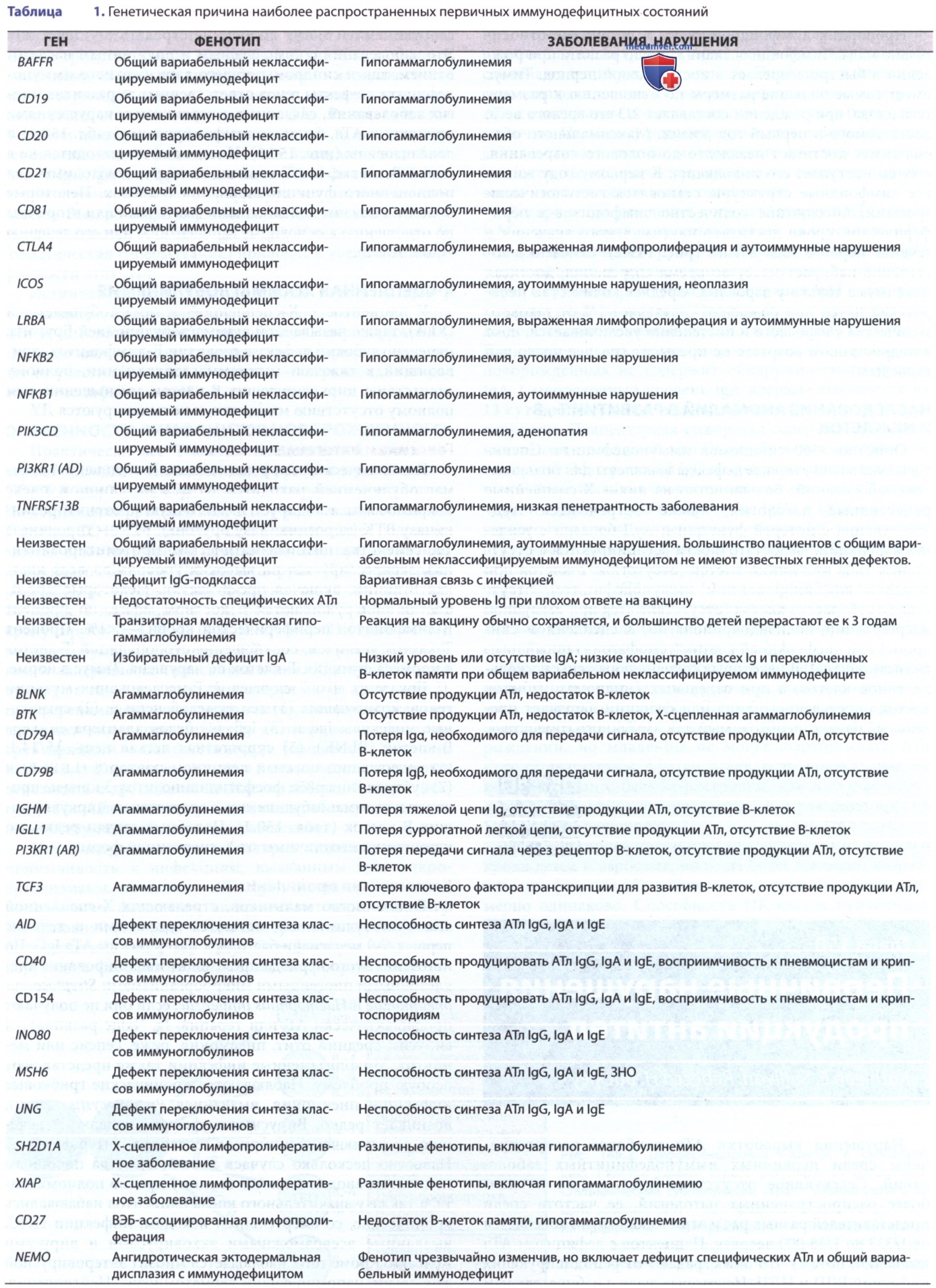
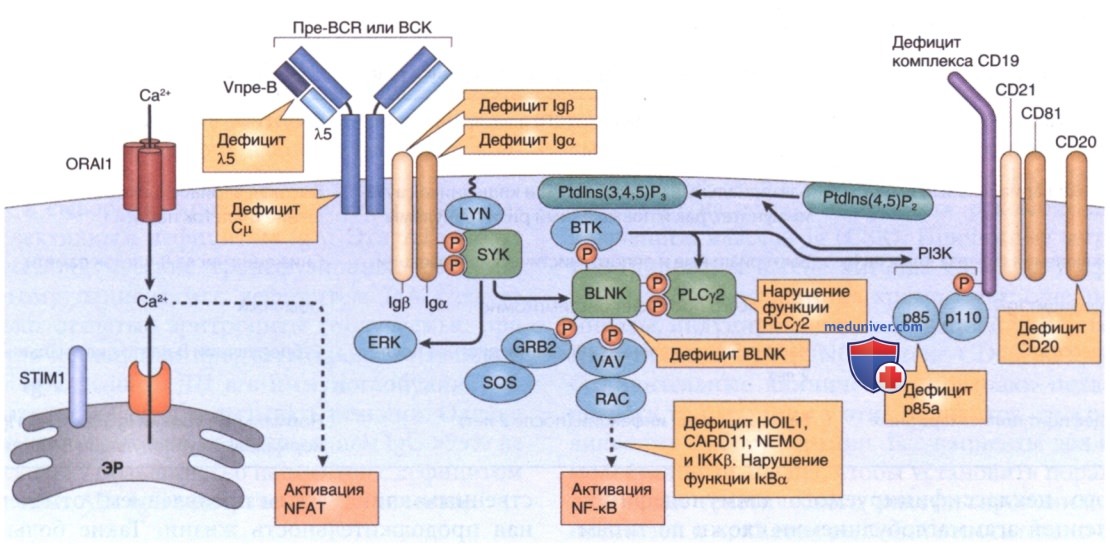
Рисунок 1. Пре-В-клетка получает сигналы пролиферации и дифференцировки через пре-В-клеточный рецептор (BCR) и корецепторы Iga и Igp. Передача сигналов от пре-BCR опосредуется иммунорецепторными тирозиновыми активирующими факторами (ITAM) корецепторов Iga и Igp, которые создают каркас и активируют тирозинкиназу SYK. SYK либо активирует внеклеточную сигнально-регулируемую киназу (ERK), либо фосфорилирует (Р) (вместе с LYN) адаптивный белок связывающий В-клетки (BLNK; англ. В-cell linker adaptor protein) и тирозинкиназу при болезни Брутона, что приводит к активации фосфолипазы Cγ2 (PLCγ2) и фосфоинозитид-3 киназы (PI3K). Нарушения этого пути передачи сказываются на npe-BCR (в Сμ или псевдолегкой цепи λ5), молекулах пре-BCR сигнальной трансдукции Igα и lgβ, расположенных ниже молекул ВТК, BLNK и PI3K, компонентах костимулирующего комплекса CD19 (CD19, CD21 и CD81) и маркере В-клеток CD20. BCR запускает конечный путь активации ядерного фактора-кВ (NF-кВ) через каркасный белок CARD11 и активацию комплекса киназы IkB (IKK) (включающего IKKa, IKKP и NEMO — основной модулятор NF-kB). Активация IKK приводит к фосфорилированию и разрушению ингибитора NF-кВ-а (1кВа) и последующему высвобождению гетеродимера р50-р65 NF-кВ, который затем перемещается в ядро для регулирования транскрипции гена (не показано). После связывания АГн со специфическими рецепторами (например, BCR) запасы Са2+ в эндоплазматическом ретикулуме истощаются, STIM1 активируется, и активируемые высвобождением Са2+ ORAI1 каналы Са2+ открываются, что приводит к управляемому из резервного депо притоку Са2*. Этот приток ведет к активации фактора транскрипции NFAT (ядерного фактора активированных Т-клеток). Пунктирные стрелки указывают сигнальные события в нисходящем направлении. ЭР — эндоплазматический ретикулум; PAD — первичный дефицит продукции АТл; Ptdlns (4,5) Р2 — фосфатидилинозитол-4,5-бисфосфат; Ptdlns (3,4,5) РЗ — фосфатидил инозитол-3,4,5-трифосфат.
а) Х-сцепленная агаммаглобулинемия. У пациентов с Х-сцепленной агаммаглобулинемией (XLA), также называемой агаммаглобулинемией Брутона, отмечен глубокий дефект в развитии В-лимфоцитов, приводящий к тяжелой гипогаммаглобулинемии, полному отсутствию циркулирующих В-клеток, уменьшению или полному отсутствию миндалин, и не пальпируются ЛУ.
1. Генетика и патогенез. Патологический ген у пациентов с Х-сцепленной агаммаглобулинемией находится на q22 на длинном плече Х-хромосомы и кодирует В-клеточную протеинтирозинкиназу ВТК (тирозинкиназу Брутона). ВТК — тирокиназа Тес-семейства цитоплазматических протеинтирозинкиназ, экспрессируется на высоких уровнях во всех клетках В-линии, включая пре-В-клетки. Некоторые пре-В-клетки обнаруживаются в костном мозге, но процент В-лимфоцитов периферической крови — <1%.
Процент Т-клеток увеличен, соотношение субпопуляций Т-клеток в норме, а функция Т-клеток не нарушена. Тимус в норме.
Доказано, что 7 случаев АуР-наследования мутаций генов, кодирующих (1) ген тяжелой цепи μ; (2) сигнальные молекулы Igα и (3) Igβ; (4) белок адаптера линкера В-клеток (BLNK); (5) суррогатная легкая цепь, λ5/14,1; (6) содержащий богатый лейцином повтор 8 (LRRC8) и (7) субъединица р85α фосфатидилинозитол-3 киназы приводят к агаммаглобулинемии с отсутствием циркулирующих В-клеток (табл. 1). Подобные случаи редки, но клинически неотличимы от Х-сцепленной формы.
2. Клинические проявления. Большинство мальчиков, страдающих Х-сцепленной агаммаглобулинемией, остаются здоровыми в течение первых 6-9 мес жизни благодаря материнским АТл IgG. По истечение этого периода происходит инфицирование внеклеточными пиогенными микроорганизмами Streptococcus pneumoniae и Haemophilus influenzae, если они не получают профилактически АБ или терапию Ig. Чаще развивается синусит, средний отит, пневмония, реже, сепсис или менингит. Микоплазменные инфекции также представляют особую проблему.
Наблюдаются хронические грибковые инфекции; пневмония, вызванная Pneumocystis jiroveci, возникает редко. Вирусные инфекции поддаются лечению, за исключением вирусов гепатита и энтеровирусов. Известно несколько случаев паралича, когда подобным пациентам вводили живую вакцину против полиомиелита, а также у значительного числа пациентов наблюдались хронические, со смертельным исходом, инфекции ЦНС, вызванные всевозможными экховирусами и вирусами Коксаки. Кроме того, наблюдается миозит энтеровирусной этиологии, напоминающий дерматомиозит. Нейтропения, выявляемая при постановке диагноза в случае инфицирования, связана с Pseudomonas или стафилококковыми инфекциями.
3. Диагностика. Диагноз Х-сцепленной агаммаглобулинемии следует предполагать, если при физикальном обследовании обнаруживается лимфоидная гипоплазия (редуцированная до минимума ткань миндалин или отсутствие пальпируемых КЕ), а сывороточные концентрации IgG, IgA, IgM и IgE намного ниже 95%-ного доверительного интервала для соответствующих (по возрасту и расе) контрольных групп; показатель общих Ig обычно <100 мг/дл.
Уровни естественных АТл к полисахаридным АГн эритроцитов типа А и В (изогемагглютининам) и АТл к АГн, полученным во время рутинной иммунизации, резко снижены при Х-сцепленной агаммаглобулинемии, но в норме при преходящей младенческой гипогаммаглобулинемии. Проточная цитометрия — важный тест для подтверждения отсутствия циркулирующих В-клеток, который позволит отличить Х-сцепленную агаммаглобулинемию от большинства типов общего вариабельного неклассифицируемого иммунодефицита, синдрома гипер-IgM и преходящей младенческой гипогаммаглобулинемии.
б) Общий вариабельный иммунодефицит. Общий вариабельный неклассифицируемый иммунодефицит — синдром, обычно сопряженный с гипогаммаглобулинемией. Уровень IgG в сыворотке не должен превышать 2 SD ниже возрастных норм, с низким уровнем IgA и IgM. Клинические проявления общего вариабельного неклассифицируемого иммунодефицита и Х-сцепленной агаммаглобулинемии схожи по типам перенесенных пациентами инфекционных заболеваний и обуславливающих их бактериальных этиологических агентов.
Исключение — энтеровирусный менингоэнцефалит, который редко встречается у пациентов с общим вариабельным неклассифицируемым иммунодефицитом (табл. 3). В отличие от Х-сцепленной агаммаглобулинемии, распределение по полу при общем вариабельном неклассифицируемом иммунодефиците почти одинаково, заболевание развивается в более позднем возрасте, а инфекции могут быть менее тяжелыми. Общий вариабельный неклассифицируемый иммунодефицит — наиболее широко распространенный гуморальный дефект.
1. Генетика и патогенез. Комбинированный вариабельный иммунодефицит — фенотипический диагноз, заболевание с полигенным наследованием. Гены, кодирующие фенотип общего вариабельного иммунодефицита после мутации, включают дефицит ICOS (индуцибельный костимулятор), SH2DIA (ответственный за Х-сцепленное лимфопролиферативное заболевание), CD19, CD20, CD21, CD81, BAFF-R (фактор активации В-клеток семейства рецепторов фактора некроза опухоли), TACI (трансмембранный активатор, модулятор кальция, и взаимодействующий циклофилин-лиганд).
Эти мутации в совокупности составляют <10% всех случаев вариабельного иммунодефицита. За редкими исключениями лечение не зависит от генетического диагноза. При атипичных инфекциях или аутоиммунных заболеваниях целесообразно проведение генетической диагностики. Некоторые генетические варианты имеют неблагоприятный прогноз: следует рассмотреть возможность трансплантации.
Несмотря на нормальное количество циркулирующих В-клеток и наличие лимфоидных кортикальных фолликулов, В-клетки крови пациентов с общим вариабельным иммунодефицитом не дифференцируются обычным путем в клетки, продуцирующие Ig. У них может наблюдаться дефицит переключаемых В-клеток памяти.
2. Клинические проявления. Дефицит сывороточного Ig и АТл у больных с общим вариабельным иммунодефицитом сопряжен с рецидивирующими синопульмональными инфекциями. Повторные легочные инфекции могут привести к развитию бронхоэктазов. Сепсис и менингит, вызванные инкапсулированными бактериями, возникают чаще, чем в общей популяции. У пациентов с рецидивирующими инфекциями (их единственным клиническим проявлением) отмечена нормальная продолжительность жизни. Такие больные хорошо переносят заместительную терапию Ig. Сопутствующие аутоиммунное заболевание или лимфопролиферация — факторы плохого прогноза.
У пациентов с общим вариабельным иммунодефицитом часто наблюдается образование ауто-АТл и нормальные или увеличенные миндалины, ЛУ; у 25% пациентов отмечена спленомегалия. Общий вариабельный иммунодефицит ассоциирован со спру-подобной энтеропатией, вызванной синдромом мальальбсорбции, которая может сопровождаться узловатой лимфоидной гиперплазией кишечника. К аутоиммунным заболеваниям относятся очаговая алопеция, гемолитическая анемия, тромбоцитопения, атрофия слизистой оболочки желудка, ахлоргидрия и злокачественная анемия.
Кроме того, наблюдаются случаи лимфоидной интерстициальной пневмонии, интерстициального заболевания легких, псевдолимфомы, В-клеточной лимфомы, амилоидоза и неказеозных, саркоидоподобных гранулем легких, селезенки, кожи и печени. Существует повышенный риск развития лимфом.
в) Селективный дефицит иммуноглобулина А (IgA). Частичное или почти полное отсутствие (<5 мг/дл) сывороточного и секреторного IgA наиболее распространено. Это выраженное иммунодефицитное расстройство, частота которого в некоторых группах населения достигает 0,33%. У пациентов возможно бессимптомное течение болезни; у них могут развиться синопульмональные или желудочно-кишечные инфекции (особенно лямблиоз). Дефицит IgA также сопряжен с целиакией и аутоиммунными заболеваниями. Диагноз не может быть поставлен до 4-летнего возраста, пока уровень IgA не достигнет уровня взрослого.
Основной генетический дефект, приводящий к дефициту IgA, неизвестен. Обнаруживаются фенотипически нормальные В-клетки крови. Подобный генетический дефект часто замечается в родословных, включающих лиц с общим вариабельным иммунодефицитом. Дефицит IgA может перерасти в общий вариабельный иммунодефицит. Дефицит IgA выявляется у пациентов, принимавших ЛП, ассоциируемые с развитием общего вариабельного иммунодефицита (фенитоин, D-пеницилламин, препараты золота и сульфасалазин). Это позволяет предположить, что факторы окружающей среды могут обуславливать развитие заболевания у генетически предрасположенного человека.
1. Клинические проявления. Инфекциям подвержены респираторный, желудочно-кишечный и урогенитальный тракт. Часто это те же бактериальные возбудители, которые вызывают инфекционные заболевания при др. синдромах дефицита АТл. Часто пациенты страдают кишечным лямблиозом. Концентрации др. Ig в сыворотке крови у пациентов с селективным дефицитом IgA в норме, хотя сообщалось о дефиците подкласса IgG2 (и др.).
АТл к IgA в сыворотке крови обнаружены у 44% пациентов с селективным дефицитом IgA. Эти АТл могут вызывать негемолитические трансфузионные реакции. Именно поэтому пациентам с дефицитом IgA следует вводить только отмытые эритроциты (получаемые при замораживании крови) или продукты крови от доноров с дефицитом IgA. Многие ЛП в/в-иммуноглобулина содержат достаточно IgA, чтобы вызывать реакции. Однако введение /в-иммуноглобулина с содержанием IgG >99% не показано, поскольку большинство пациентов с дефицитом IgA вырабатывают АТл IgG в достаточном количестве.
г) Дефицит иммуноглобулина G-подкласса. У некоторых пациентов, несмотря на нормальную или повышенную концентрацию общего IgG в сыворотке крови, наблюдается дефицит одного или нескольких из 4 подклассов IgG. Иногда у пациентов с отсутствием или очень низкой концентрацией IgG2 также обнаруживается дефицит IgA. У др. пациентов с дефицитом подкласса IgG развивается общая вариабельная иммунная недостаточность. Это позволяет предположить, что дефицит IgG-подкласса может быть маркером общей иммунной дисфункции. Биологическое значение наблюдаемых многочисленных умеренных дефицитов подклассов IgG трудно оценить.
Количественное определение подкласса IgG нецелесообразно при оценке иммунной функции у ребенка с рецидивирующей инфекцией. Более актуальная задача — способность пациента вырабатывать специфические АТл к белковым и полисахаридным АГн, потому что глубокий дефицит антиполисахаридных АТл отмечен даже при нормальных концентрациях IgG2. В/в-иммуноглобулин не следует вводить пациентам с дефицитом IgG-подкласса, если у них не выявлен дефицит АТл к широкому спектру АГн.
д) Делеции тяжелых и легких цепей иммуноглобулинов. У некоторых пациентов с бессимптомным проявлением заболевания зарегистрировано полное отсутствие IgG1, IgG2, IgG4 и IgA1 в результате делеций генов. Этот факт подтверждает важность исследования образования специфических АТл перед принятием решения о начале терапии в/в-иммуноглобулином у пациентов с дефицитом IgG-подкласса.
е) Транзиторная младенческая гипогаммаглобулинемия. Транзиторная гипогаммаглобулинемия обнаруживается у младенцев при лабораторных исследованиях, свидетельствуя о задержке синтеза Ig. Она встречается у 1:1000 детей. Большинство младенцев начинают вырабатывать IgG в первые 3 мес жизни, и их количество увеличивается на протяжении младенчества. По непонятным причинам у небольшого количества младенцев этот процесс начинается поздно или недостаточно продуктивен. Это состояние разрешается без вмешательства, но затрудняет диагностику.
Ключевое различие — в этом состоянии ответы на вакцины сохраняются, а в др. случаях иммунный ответ снижен или отсутствует.
ж) Дефект переключателя класса иммуноглобулинов. Синдром гипер-IgM — генетически гетерогенный, характеризуется нормальным или повышенным уровнем сывороточного IgM, связанным с низким уровнем или отсутствием сывороточных уровней IgG, IgA и IgE, что указывает на дефект в процессе рекомбинации с переключением классов Ig (CSR). Причинные мутации идентифицированы в гене лиганда CD40 на Х-хромосоме и трех генах на аутосомных хромосомах: гене цитидиндезаминазы, индуцированной активацией (AID), гене урацил-ДНК-гликозилазы (UNG) и гене CD40 на хромосоме 20.
Отличительные клинические признаки позволяют распознать тип мутации у этих пациентов, способствуя правильному выбору терапии. Все пациенты должны пройти молекулярный анализ, чтобы установить пораженный ген для целей генетического консультирования, выявления носителей и принятия решений относительно окончательной терапии.
1. Х-сцепленный гипериммуноглобулин М, вызванный мутациями в гене лиганда CD40. Х-сцепленный гипер-IgM вызван мутациями в гене, кодирующем лиганд CD40 (CD154, CD40L), который экспрессируется на активированных Т-хелперных (Th) клетках. У мальчиков с этим синдромом отмечены очень низкие сывороточные концентрации IgG и IgA с нормальной или повышенной концентрацией поликлональных IgM. У пациентов отмечена гипоплазия миндалин, ЛУ обычно не пальпируются; часто наблюдается глубокая нейтропения.
- Генетика и патогенез. В-клетки не имеют патологических изменений при этом заболевании; дефект обнаруживается в Т-клетках. CD40L — лиганд для CD40, который находится на В-клетках и моноцитах. CD40L активируется на активированных Т-клетках. Мутации приводят к неспособности сигнализировать В-клеткам о переключении изотипа. Таким образом, В-клетки продуцируют только IgM. Неспособность Т-клеток взаимодействовать с В-клетками через эту пару рецептор-лиганд также приводит к неспособности позитивной регуляции поверхностных молекул В-клеток и моноцитов CD80 и CD86, которые взаимодействуют с CD28/CTLA4 (цитотоксического Т-лимфоцитарного антигена 4) на Т-клетках, что приводит к неспособности перекрестного взаимодействия между клетками иммунной системы.
- Клинические проявления. Как и у пациентов с Х-сцепленной агаммаглобулинемией, у мальчиков с дефектом лиганда CD40 в течение 1-2 лет жизни развиваются рецидивирующие гнойные инфекции: средний отит, синусит, пневмония и тонзиллит. Они имеют выраженную предрасположенность к пневмонии, вызванной Р. jirovecii, и риск развития нейтропении. Гистология ЛУ показывает только абортивное формирование зародышевого центра с серьезным истощением и фенотипическими аномалиями фолликулярных дендритных клеток. Уровень циркулирующих В-лимфоцитов у этих пациентов находится в норме, но частота CD27+ В-клеток памяти снижена. Циркулирующие Т-клетки также присутствуют в нормальном количестве, и ответы на митогены in vitro нормальны, но наблюдается снижение АГн-специфической функции Т-клеток.
Помимо условно-патогенных инфекций (пневмония, вызванная Р. Jirovecii), часто встречаются множественные вульгарные бородавки (verruca vulgaris), энтерит, вызванный Cryptosporidium, дальнейшее поражение печени и повышенный риск ЗНО.
- Лечение. Из-за плохого прогноза предпочтительный метод лечения — трансплантация HLA-идентичных гемопоэтических стволовых клеток, предпринятая в раннем возрасте. В качестве альтернативного лечения может проводиться ежемесячная инфузия в/в-иммуноглобулина. Пациентам с тяжелой формой нейтропении рекомендовано использование гранулоцитарного колониестимулирующего фактора.
2. Аутосомно-рецессивный гипериммуноглобулин М:
- Генетика и патогенез. В отличие от пациентов с дефектом CD40L, В-клетки этих пациентов не могут переключаться с IgM-секретирующих клеток на IgG-, IgA- или IgE-секретирующие клетки, даже при совместном культивировании с нормальными Т-клетками. Все дефекты относятся ко внутренними дефектами В-клеток. Наиболее часто наблюдается АуР-дефект гена, кодирующего AID. В целевой ДНК AID дезаминирует цитозин, превращая его в урацил, после чего UNG удаляет урацил. Серьезный дефект CSR обнаружен у 3 пациентов с гипер-IgM-синдромом, сообщивших что им был диагностирован дефицит UNG. Клинические особенности аналогичны таковым при дефиците AID с повышенной восприимчивостью к бактериальным инфекциям и лимфоидной гиперплазии.
Гистологическое исследование увеличенных ЛУ показало наличие гигантских зародышевых центров (в 5-10 раз больше нормы), заполненных высокопролиферирую-щими В-клетками. АуР-гипер-IgM может быть вызван дефектами CD40. Клинические проявления включали рецидивирующие синусно-легочные инфекции, пневмонию, вызванную Р. jirovecii, и инфекции Cryptosporidium parvum, очень похожие на симптомы, наблюдаемые при синдроме Х-сцепленного гипер-IgM.
- Клинические проявления. У больных с дефицитом AID, UNG и CD40 наблюдаются низкие концентрации сывороточных IgG, IgA и IgE. Сывороточная концентрация IgM у пациентов с дефицитом AID заметно повышена и поликлональна, что не обнаруживается при дефекте CD40 лиганда. Пациенты с мутациями AID и UNG, страдающие лимфоидной гиперплазией, старше по возрасту в момент начала болезни, не предрасположены к пневмонии, вызванной Р. jirovecii, часто имеют изогемагглютинины. У них гораздо реже встречается нейтропения, если она не возникает на аутоиммунной основе. Такие пациенты склонны к развитию аутоиммунных и воспалительных заболеваний, включая СД, полиартрит, аутоиммунный гепатит, гемолитическую анемию, иммунную тромбоцитопению, болезнь Крона и хронический увеит.
- Лечение и прогноз. При ранней диагностике и ежемесячных введениях Ig, при адекватном лечении инфекций с помощью АБ у пациентов с мутациями AID и UNG наблюдается более доброкачественное течение, чем у мальчиков с дефектами CD40L или CD40. Дефицит CD40 встречается редко, но точно имитирует проявления CD40L.
3. Х-сцепленный лимфопролиферативный синдром. Известно 2 типа Х-сцепленного лимфопролиферативного синдрома (табл. 4). У каждого из них есть отличительные клинические признаки, но они обуславливают одинаковую восприимчивость к ВЭБ и развитию гемофагоцитарного лимфогистиоцитоза (HLH; англ. hemophagocytic lympho-histiocytosis).
- Генетика и патогенез. Патологический ген при Х-сцепленном лимфопролиферативном синдроме типа 1 был локализован на Xq25, клонирован; продукт гена изначально назван SAP (SLAM-ассоциированный белок), но теперь ему присвоено официальное название SH2D1A. SLAM (сигнальная молекула активации лимфоцитов) — молекула адгезии, которая активируется как на Т-, так и на В-клетках в случае инфекционных заболеваний или др. стимуляции. Отсутствие SH2D1A может привести к развитию неконтролируемого цитотоксического Т-клеточного иммунного ответа на ВЭБ. В естественных условиях белок SH2D1A избирательно связывается с рецептором 2В4 на NK-клетках (естественные клетки-киллеры).
Избирательное нарушение 2В4-опосредованной активации NK-клеток также играет немаловажную роль в иммунопатологии Х-сцепленного лимфопролиферативного синдрома. Х-сцепленный лимфопролиферативный синдром типа 2 вызван мутацией XIAP (Х-сцепленный ингибитор белка апоптоза). Проявления заболевания аналогичны симптомам Х-сцепленного лимфопролиферативного синдрома. Точная роль этого белка в формировании восприимчивости к ВЭБ не выяснена.
- Клинические проявления. Мужчины - носители мутации здоровы до заражения ВЭБ. Средний возраст проявления симптомов — <5 лет. Выделяют 3 основных клинических фенотипа: (1) фульминантный, часто смертельный инфекционный мононуклеоз (50% случаев); (2) лимфомы, преимущественно В-клеточные (25%); и (3) приобретенная гипогаммаглобулинемия (25%). Менее распространенное проявление — васкулит ЦНС. Отмечается заметное нарушение выработки АТл к ядерному АГн ВЭБ, тогда титры АТл к вирусному капсидному АГн варьируют от отсутствующих до значительно повышенных.
При Х-сцепленном лимфопролиферативном синдроме прогноз неблагоприятный. За исключением случаев наличия Х-сцепленного лимфопролиферативного синдрома в семейном анамнезе, диагностика этого заболевания до появления осложнений затруднена, поскольку изначально болезнь протекает бессимптомно.
Сообщается, что в 2 родословных у мальчиков в одной ветви из каждой родословной диагностирован общий вариабельный иммунодефицит, а мальчики из др. ветви страдали острым инфекционным мононуклеозом. Члены семьи с общим вариабельным иммунодефицитом никогда не страдали инфекционным мононуклеозом. Все вовлеченные члены каждой родословной имели одинаковую мутацию SH2D1A, несмотря на различие клинических фенотипов. Поскольку мутация SH2D1A одинакова, а фенотип в этих семьях варьировал, возможность развития Х-сцепленного лимфопролиферативного синдрома следует рассматривать у всех мужчин с диагнозом общий вариабельный иммунодефицит, особенно при наличии >1 члена семьи мужского пола с этим фенотипом.