Заболевания, связанные с первичным иммунодефицитом, характеризуются нарушением регуляции иммунитета, аутоиммунитетом и аутоиммунным воспалением относятся к моногенным дефектам иммунной системы. Эти сложные полисистемные заболевания характеризуются прогрессирующим фенотипом с органоспецифическим аутоиммунитетом, специфической инфекционной восприимчивостью и лимфопролиферацией.
а) Аутоиммунный лимфопролиферативный синдром. Аутоиммунный лимфопролиферативный синдром, также известный как синдром Канале-Смита, — нарушение апоптоза лимфоцитов, приводящее к поликлональным популяциям Т-клеток (дважды «-» Т-клетки), которые экспрессируют рецепторы АГн CD3 и α/β, но не содержат ко-рецепторов CD4 или CD8 (CD3+ Т-клеточный рецептор α/β +, CD4-CD8-).
Эти Т-клетки плохо реагируют на АГн или митогены и не продуцируют факторы роста или выживания (IL-2). Генетический дефицит у большинства пациентов представляет зародышевую или соматическую мутацию в гене FAS, который продуцирует рецептор на клеточной поверхности суперсемейства рецепторов ФНО (TNFRSF6), который при стимуляции его лигандом вызывает запрограммированную гибель клеток (табл. 3). Устойчивое выживание этих лимфоцитов приводит к нарушению регуляции иммунитета и аутоиммунитету. Аутоиммунный лимфопролиферативный синдром также вызывается др. генами в пути Fas (FASLG и CASP10).
Кроме того, подобные аутоиммунному лимфопролиферативному синдрому нарушения связаны с др. мутациями: RAS-ассоциированным аутоиммунным лимфопролиферативным расстройством (RALD; англ. RAS-associated autoimmune lymphoproliferative disorder), дефицитом каспазы-8, Fas-ассоциированным белком с дефицитом домена смерти (FADD; англ. Fas-associated protein with death domain deficiency) и дефицитом дельта протеинкиназы С (PRKCD; англ. protein kinase С delta deficiency). Эти расстройства сопровождаются разной степенью иммунодефицита, аутоиммунитета и лимфопролиферации.
1. Клинические проявления. Аутоиммунный лимфопролиферативный синдром характеризуется аутоиммунитетом, хронической персистирующей или рецидивирующей лимфаденопатией, спленомегалией, гепатомегалией (в 50%) и гипергаммаглобулинемией (IgG, IgA).
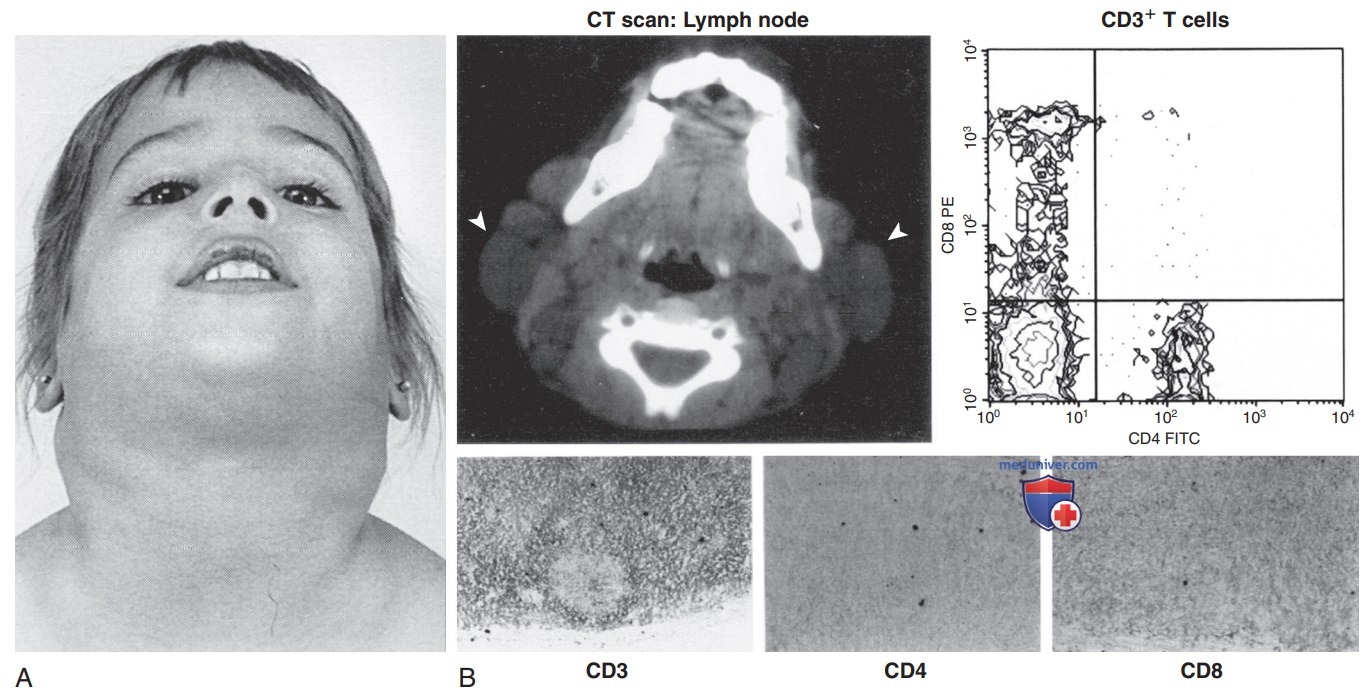
Многие пациенты обращаются к врачу на первом году жизни, но у большинства симптомы появляются к 5 годам. Лимфаденопатия в некоторых случаях имеет ярко выраженные проявления (рис. ниже). Спленомегалия может привести к гиперспленизму. Аутоиммунитет — причина анемии (Кумбс-«+» гемолитическая анемия), тромбоцитопении или легкой степени нейтропении. Лимфопролиферативный процесс (лимфаденопатия, спленомегалия) может со временем регрессировать, но аутоиммунитет не регрессирует и характеризуется частыми обострениями и рецидивами. Др. аутоиммунные симптомы включают крапивницу, увеит, гломерулонефрит, гепатит, васкулит, панникулит, артрит и поражение ЦНС (судороги, головные боли, энцефалопатию).
Клинические, рентгенографические, иммунологические и гистологические характеристики аутоиммунного лимфопролиферативного синдрома. Примечание. (А) Вид спереди пациента Национального института здоровья США. (B) Вверху в середине КТ шеи показывает увеличенные преаурикулярные, шейные и затылочные ЛУ. Стрелки обозначают наиболее выступающие ЛУ. Верхние правые панели показывают проточно-цитометрический анализ Т-клеток периферической крови пациента с аутоиммунным лимфопролиферативным синдромом с экспрессией CD8 по вертикальной оси и CD4 по горизонтальной оси. Нижний левый квадрат содержит CD4-CD8- (дважды «-») Т-клетки, которые присутствуют в <1% Т-клеток, экспрессирующих Т-клеточный рецептор αβ. На нижних панелях показано окрашивание CD3, CD4 и CD8 на серийных срезах образца биопсии ЛУ от пациента с аутоиммунным лимфопролиферативным синдромом. Также показано, что большое количество DNCD3+ CD4- CD8- (дважды «-») Т-клеток находятся в межфолликулярных областях лимфатического узла.
ЗНО чаще встречаются у пациентов с аутоиммунным лимфопролиферативным синдромом. К ним относят лимфомы Ходжкина и неходжкинские лимфомы, опухоли плотных тканей ЩЖ, кожи, сердца или легких. Аутоиммунный лимфопролиферативный синдром — одна из причин синдрома Эвана (иммунная тромбоцитопения и иммунная гемолитическая анемия).
2. Диагностика. Отклонения от нормы, выявленные путем лабораторных исследований, зависят от реакции лимфопролиферативного органа (гиперспленизм) или степени аутоиммунитета (анемия, тромбоцитопения). Возможно развитие лимфоцитоза или лимфопении. В табл. 3 приведен список ДК. Проточная цитометрия помогает определить тип лимфоцитов (рис. выше). Функциональный анализ гена TNFRSF6 часто выявляет гетерозиготную мутацию.
3. Лечение. Рапамицин (сиролимус) часто назначается для контроля проявлений аденопатии и аутоиммунных цитопений. ЗНО лечат с помощью обычных протоколов, используемых у пациентов, не страдающих аутоиммунным лимфопролиферативным синдромом. Трансплантация стволовых клеток — один из возможных вариантов лечения аутоиммунных проявлений.
б) Иммунная дисрегуляция, полиэндокринопатия, энтеропатия, Х-сцепленный синдром. Синдром иммунной дисрегуляции начинается в течение первых нескольких недель или месяцев жизни. Характерные проявления: водянистая диарея (аутоиммунная энтеропатия), экзематозная сыпь (эритродермия у новорожденных), инсулинозависимый СД, гипертиреоз или гипотиреоз, тяжелая аллергия и др. аутоиммунные нарушения (Кумбс-«+» гемолитическая анемия, тромбоцитопения, нейтропения).
Также сообщалось о псориазиформной или ихтиозиформной сыпи и облысении. Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии, энтеропатии (IPEX-синдром) вызван мутацией в гене FOXP3, который кодирует фактор транскрипции скурфин, участвующий в функционировании и развитии регуляторных Т-клеток CD4+ CD25+ (T-reg). Отсутствие T-reg может предрасполагать к патологической активации эффекторных Т-клеток. Доминирующие мутации, связанные с приобретением функции STAT1, и мутации др. генов (табл. 4) вызывают синдром, подобный IPEX, также связанный с нарушением T-reg.
1. Клинические проявления. Водянистая диарея с атрофией кишечных ворсинок приводит к задержке развития у большинства пациентов. Поражения кожи (экзема) и инсулинозависимый диабет начинаются в младенчестве, также обнаруживаются лимфаденопатия и спленомегалия. Серьезные бактериальные инфекции (менингит, сепсис, пневмония, остеомиелит) могут быть связаны с нейтропенией, недостаточностью питания или иммунной дисрегуляцией. Результаты лабораторных исследований отражают сопряженные с ними аутоиммунные заболевания, обезвоживание и недоедание. Уровни IgE в сыворотке повышены при нормальных уровнях IgM, IgG и IgA. Диагноз ставится клинически и путем анализа мутаций гена FOXP3.
2. Лечение. Подавление активации Т-клеток циклоспорином, такролимусом или сиролимусом с ГКС — лечение первой линии, наряду со специфическим лечением эндокринопатии и др. проявлений аутоиммунитета. Эти агенты используются в качестве моста к трансплантации. Трансплантация гемопоэтических стволовых клеток — единственная возможность лечения IPEX-синдрома.
в) Дефицит цитотоксического Т-лимфоцитарного антигена 4 (CTLA4). Пациенты с дефицитом CTLA4 утратили способность поддерживать иммунную толерантность. Характерные проявления: аутоиммунитет и полиорганная лимфоцитарная инфильтрация лимфоидных и нелимфоидных органов. АГн CTLA4, также известный как CD152, — рецептор белка, экспрессируемого на активированных Т-клетках. Он действует как иммунная контрольная точка, подавляя иммунные реакции на активацию Т-клеток. Дефицит CTLA4 наследуется по гаплонедостаточному типу.
Основные проявления: аутоиммунные цитопении, лимфоидная инфильтрация лимфоидных и нелимфоидных органов, гранулематозная болезнь, гипогаммаглобулинемия и рецидивирующие респираторные инфекции. Нелимфоидные органы, чаще всего затронутые лимфоидной инфильтрацией: ГМ и ЖКТ. Иммунный фенотип пациентов с дефицитом CTLA4 включает снижение наивных Т-клеток (CD4+ CD45RA+ D62L+), потерю циркулирующих В-клеток и снижение экспрессии T-reg. Лечение симптоматическое, хотя использование абатацепта, гибридного белка CTLA4-Ig, облегчило симптомы заболевания у нескольких пациентов. При невосприимчивости к терапии оправдана трансплантация гемопоэтических стволовых клеток, которая приводит к ремиссии и полному исчезновению симптомов.
г) Дефицит липополисахарид-связывающего белка (LRBA). Гомозиготные мутации гена LRBA — причина синдрома гипогаммаглобулинемии, характеризующегося ранним началом, аутоиммунитетом, лимфопролиферацией и воспалительным заболеванием кишечника. Белок LRBA — член семейства протеинов PH-BEACH-WD40 (англ. Pleckstrin homology-beige and Chediak-Higashi-tryptophan-aspartic acid dipeptide). О функции LRBA известно немногое. В нормальных Т-клетках LRBA располагается совместно с CTLA4 в рециклирующих эндосомах: это позволяет предположить, что LRBA играет специфическую роль в регуляции рециркуляции эндосом. Гомозиготные мутации в LRBA останавливают экспрессию белка LRBA.
Наиболее частые проявления иммунной дисрегуляции: энтеропатии, аутоиммунная цитопения, гранулематозно-лимфоцитарное интерстициальное заболевание легких, лимфаденопатия, гепатомегалия или спленомегалия. Менее распространенные проявления иммунной дисрегуляции: церебральные гранулемы, СД-1, алопеция, увеит, миастения и экзема. У многих пациентов наблюдается задержка роста, осложненная энтеропатией. Бактериальные, грибковые и вирусные инфекции диагностированы у 50% пациентов. Иммунный фенотип вариабелен, но может состоять из пониженного количества Т-клеток (CD3+), повышенного количества дважды «-» Т-клеток (CD3+ CD4-CD8-), нормальной пролиферации Т-клеток на митогены и АГн, пониженного числа T-reg (CD4+ CD25+ FoxP3+), маленьких NK-клеток (CD56+) и маленьких В-клеток (CD19+). Количество иммуноглобулинов также варьирует, часто обнаруживается гипогаммаглобулинемия.
При назначении терапии основное внимание уделяется лечению иммунодисрегуляторных проявлений с помощью иммуносупрессии. ГКС, заместители иммуноглобулина, микофенолата мофетил, такролимус, рапамицин, будесонид, циклоспорин, азатиоприн, ритуксимаб, инфликсимаб и гидроксихлорохин применялись с переменным успехом. Абатацепт успешно применялся для лечения иммунодисрегуляторные проявлений. Трансплантация гемопоэтических стволовых клеток успешно проводилась у пациентов с дефицитом LRBA.
д) Синдромы активированной фосфоинозитид-3-киназы Δ (PI3K). Эти синдромы относятся к первичным иммунодефицитам, которые вызывают лимфаденопатии и старение Т-клеток. Молекулы PI3K состоят из каталитической субъединицы р110 (р110α, р110β или р110δ) и регуляторной субъединицы (р85α, р55α, р50α, р85β или р55γ). PI3K превращают фосфатидилинозитол-4,5-бисфосфат в фосфатидилиноситол-3,4,5-трифосфат (PIP 3), важный второй мессенджер.
АуД-мутации с усилением функции в PIK3CD, гене, который кодирует каталитическую единицу, p110δ, приводит к гиперактивированной передаче сигналов PI3Kδ. АуД-мутации в PI3KR, гене, кодирующем регуляторную субъединицу (р85α, р55α и р50α) PI3K, ассоциируются с тем же фенотипом. Нарушения этого пути приводят к синдрому хронической лимфопролиферации и старения Т-клеток.
Наиболее частые симптомы: ранние инфекции ДП, неинфекционная лимфаденопатия и гепатоспленомегалия. В результате рецидивирующей пневмонии у большой части пациентов в раннем возрасте развиваются бронхоэктазы. Также распространены стойкие, тяжелые или рецидивирующие герпесвирусные инфекции. Лимфаденопатия часто начинается в детстве и локализуется в очагах инфекции. Лимфаденопатия может быть диффузной и чаще связана с хронической ЦМВ- или ВЭБ-виремией. Также часто встречается лимфоидная гиперплазия слизистой оболочки ДП и ЖКТ. Гистологический анализ ЛУ выявляет атипичную фолликулярную гиперплазию. Аутоиммунные цитопении — наиболее частые аутоиммунные проявления; др. симптомы: гломерулопатии, опосредованные ауто-АТл, заболевание ЩЖ и склерозирующий холангит.
Сообщалось также о раннем проявлении лимфомы, возникающей уже на втором году жизни (основная причина смертности). Также наблюдается нарушение роста и прибавки МТ с задержкой психомоторного развития и умеренными когнитивными нарушениями.
Иммунофенотип состоит из пониженного количества наивных Т-клеток (CD3+ CD4+) и количества В-клеток (CD19+) при нормальном содержании NK-клеток (CD56+). Характерно снижение числа недавних «эмигрантов» из тимуса (CD3+ CD+ CD45RA+ CD31+) с повышенным количеством цитотоксических Т-клеток памяти эффекторов (CD3+ CD8+ CCR7- CD45RA+/-), увеличение переходных В-клеток (CD19+ IgM+ CD38+), уменьшение непереключаемых В-клеток памяти (CD 19+ IgD+ CD27+) и В-клеток памяти с переключением классов (CD19+ IgD+ CD27+). Уровни иммуноглобулина варьируют, но наблюдается повышенное количество IgM в сыворотке, пониженное или нормальное количество IgG и IgA.
Лечение симптоматическое, но может включать антимикробную профилактику и заместительную терапию иммуноглобулинами. Различные иммунодепрессанты (например, ритуксимаб, рапамицин) использовались для лечения часто сопутствующих лимфопролиферативных заболеваний и аутоиммунных цитопений. Трансплантация гемопоэтических стволовых клеток успешно проводилась у пациентов, невосприимчивых к медикаментозной терапии.
е) Нарушение пути сигнального преобразователя и активатора транскрипции (STAT). Путь сигнала янус-киназ (JAK-STAT) используется для передачи сигнала цитокиновыми рецепторами типа 1 и 2 в большинстве гемопоэтических клеток. Цитокины связываются со специфическим рецептором, активируя сигнальные пути JAK-STAT, что приводит к активации генов, участвующих в иммунном ответе на многие патогены. Существует 4 белка JAK (Jak1, Jak2, Jak3, Tyk2) и 6 STAT (1-6). Мутации в нескольких JAK и STAT — причина иммунодефицита. В табл. 5 рассмотрены заболевания, влияющие на белки STAT и характеризующиеся нарушением иммунной регуляции.
Хроническая иммуносупрессия необходима для контроля дефектов STAT. Руксолитиниб, ингибитор JAK-STAT, использовался с некоторым успехом. С появлением иммуномодулирующей терапии JAK-STAT пациентам будет предложено больше вариантов лечения.
ж) Нарушения пути ядерного фактора-КВ. Пути NF-кВ состоят из канонических (NF-кВ1) и неканонических (NF-kB2) путей. При клеточной активации оба пути приводят к активации и транслокации белков NF-кВ в ядро, где они инициируют последующие воспалительные реакции. В табл. 6 описаны иммунные дефекты путей NF-кВ, которые вызывают симптомы иммунной дисрегуляции или аутоиммунитета. Лечение дефектов NF-кВ включает профилактику инфекций и замену иммуноглобулина, а также трансплантацию гемопоэтических стволовых клеток.
з) Дефицит тетра-трикопептидного домена 7А (TTC7A). Комбинированный иммунодефицит с дефектами Т-клеток и В-клеток долгое время сопровождался наследственной множественной атрезией кишечника. Мутации в ТТС7А — причина сочетания кишечных и иммунологических дефектов. ТТС7А участвует в контроле клеточного цикла, организации цитоскелета, форме и полярности клеток, в клеточной адгезии. Дефицит ТТС7А наследуется по АуР-типу. Множественная атрезия кишечника с нарушением архитектуры кишечника — универсальный признак. Часто одновременно возникает тяжелый энтероколит с ранним началом. Описан иммунодефицит с тяжелой Т-клеточной лимфопенией; В- и NK-клеточные дефекты вариабельны. Пролиферативные ответы Т-клеток также ненормальны. Часто встречается тяжелая гипогаммаглобулинемия.
Лечение состоит в удалении атретических участков кишечника и антимикробной профилактики у пациентов с иммунодефицитом. Пересадка кишечника также может быть успешной.
и) Дефицит аденозиндезаминазы-2 (DADA2). Дефицит аденозиндезаминазы-2 — причина ранней васкулопатии, инсульта и иммунодефицита. Дефицит аденозиндезаминазы-2 вторичен по отношению к АуР-мутациям в области 1-й хромосомы синдрома кошачьего глаза (CECR1), картированной на хромосоме 22q11.1. Дефицит аденозиндезаминазы-2 играет важную роль в метаболизме пуринов, превращая аденозин в инозин и 2’-дезоксиаденозин в 2’-дезоксиинозин. Патогенез точно не известен, но аденозиндезаминаза-2 в основном секретируется миелоидными клетками, а дефицит приводит к усилению регуляции провоспалительных генов и повышенной секреции провоспалительных цитокинов.
Дефицит аденозиндезаминазы-2 характеризуется хроническим или рецидивирующим воспалением с повышенной реактивностью острой фазы и лихорадкой. Кожные проявления включают сетчатое ливедо, макулопапулезную сыпь, узелки, пурпуру, узловатую эритему, феномен Рейно, язвенные поражения и некроз пальцев. Поражение ЦНС варьируется, возможны преходящие ишемические атаки и ишемический или геморрагический инсульт. Периферическая невропатия встречается часто. Проявления со стороны ЖКТ: гепатоспленомегалия, гастрит, перфорация кишечника и портальная гипертензия. Нефрогенная гипертензия встречается часто и может быть связана с гломерулосклерозом или амилоидозом. Иммунодефицит проявляется гипогаммаглобулинемией и вариабельным снижением уровня IgM.
Длительное лечение ГКС и препаратами против ФНО-α показало умеренный контроль над проявлениями заболевания. Трансплантация гемопоэтических стволовых клеток успешно проведена у 2 больных.