Талассемия относится к группе генетических нарушений производства глобиновых цепей, при которых наблюдается дисбаланс между производством α- и β-глобиновых цепей. Синдромы β-талассемии возникают в результате снижения количества цепей β-глобина, что приводит к относительному избытку цепей α-глобина. При β0-талассемии производство β-глобина отсутствует. Если пациенты гомозиготны по гену β0-талассемии, их организм не может производить нормальные цепи β-глобина (HbА).
β+-талассемия указывает на мутацию, вызывающую снижение уровня нормального β-глобина (HbА). Синдромы β0-талассемии обычно протекают в более тяжелой форме, чем синдромы β+-талассемии, но имеет место значительная вариабельность между генотипом и фенотипом. Большая β-талассемия/трансфузионно-зависимая талассемия относится к тяжелой форме, требующей ранней гемотрансфузионной терапии.
Промежуточная β-талассемия/не зависимая от гемотрансфузий — менее тяжелый клинический фенотип, который обычно не требует регулярной гемотрансфузионной терапии в детском возрасте. Многие из этих пациентов имеют, по крайней мере, одну мутацию β+-талассемии. Синдромы β-талассемии обычно подразумевают мутации β-талассемии в обоих генах β-глобина. Носители с единственной мутацией β-глобина симптомов обычно не имеют, за исключением микроцитоза и легкой анемии.
При α-талассемии наблюдается отсутствие/снижение производства α-глобина, как правило, вследствие делеции генов α-глобина. ЗЛ имеют 4 гена α-глобина; чем больше генов поражено, тем тяжелее заболевание. α0-талассемия указывает на отсутствие α-цепей, продуцируемых этой хромосомой (--/). При α+-талассемии из этой хромосомы синтезируется сниженное количество цепей α-глобина (-альфа/).
Первичная патология при синдромах талассемии обусловлена количеством производимого глобина, тогда как первичная патология при СКБ зависит от качества производимого β-глобина.
а) Эпидемиология. Существует >200 разл. мутаций, приводящих к отсутствию/снижению производства глобина. Хотя большинство из них редко встречаются, 20 наиболее распространенных аномальных аллелей составляют 80% известных талассемий во всем мире; 3% населения мира являются носителями аллелей β-талассемии, а в Юго-Восточной Азии 5-10% населения являются носителями аллелей α-талассемии. В отдельно взятом регионе встречается меньше общих аллелей. В США, по оценкам, 2000 человек страдают большой β-талассемией.
б) Патофизиология. Два родственных признака оказывают влияние на развитие осложнений при синдромах β-талассемии: неадекватное производство генов β-глобина, приводящее к снижению уровня нормального HbА, и несбалансированное производство цепей α- и β-глобина, приводящее к неэффективному эритропоэзу. При β-талассемии α-глобиновые цепи находятся в избытке по сравнению с не-α-глобиновыми цепями, а сформированные тетрамеры α-глобина (α4) появляются в виде включений в эритроцитах. Свободные α-глобиновые цепи и включения очень нестабильны, накапливаются в клетках-предшественниках эритроцитов, повреждают мембрану эритроцитов и сокращают время их жизни, что приводит к анемии и увеличению производства эритроидов (табл. 7).
Это вызывает заметное увеличение эритропоэза с ранней гибелью эритроидных предшественников в костном мозге. Клинические характеристики представлены отсутствием созревания эритроцитов и неадекватно низким количеством ретикулоцитов. Такой неэффективный эритропоэз и компенсаторное массивное расширение костного мозга с эритроидной гиперактивностью характерны для β-талассемии. Из-за низкого производства β-глобина/его отсутствия α-цепи соединяются с γ-цепями, в результате чего HbF (α2γ2) становится доминирующим.
В дополнение к естественной продолжительности жизни, цепи γ-глобина м.б. произведены в большем, чем необходимо, количестве, регулируемом генетическими полиморфизмами.
Синтез δ-цепи при β-талассемии/малой β-талассемии обычно не затрагивается, и поэтому пациенты имеют относительное/абсолютное повышение уровней производства HbА2 (α2δ2).
При синдромах α-талассемии наблюдается снижение производства α-глобина. В норме существует 4 гена α-глобина (по 2 от каждого родителя), которые контролируют его выработку. Синдромы α-талассемии варьируют от полного отсутствия (отечный синдром новорожденных) до незначительно сниженного (α-талассемия бессимптомного носителя) производства α-глобина. При синдромах α-талассемии образуется избыток β- и γ-глобиновых цепей. Эти избыточные цепи формируют Hb Барта (γ4) во время фетального периода жизни плода и HbН (β4) после рождения.
Эти аномальные тетрамеры представляют собой нефункциональные Hb с очень высоким сродством к кислороду. Они не транспортируют кислород, и в результате возникает внесосудистый гемолиз. У плода с наиболее тяжелой формой α-талассемии (отечным синдромом новорожденных) развивается в/утробная анемия, и беременность обычно заканчивается выкидышем, поскольку для производства HbF требуется достаточного количества α-глобина. В то же время у детей с большой β-талассемией симптомы проявляются только после рождения, когда начинает преобладать HbА, и недостаточное производство β-глобина проявляется клинически.
1. Клинические проявления. Без лечения у детей с гомозиготной β0-талассемией обычно появляются симптомы прогрессирующей анемии с сильной слабостью и сердечной декомпенсацией в период с 6 мес до года. В зависимости от мутации и степени выработки фетального Hb необходимы регулярные гемотрансфузии, начиная со 2-го мес до 2-го года жизни, но они редко требуются в более старшем возрасте. Принятие решения о проведении гемотрансфузии зависит от многих факторов, но не обуславливается исключительно степенью анемии.
Признаки неэффективного эритропоэза, такие как отставание в росте, деформации костей как следствие расширения костного мозга, гепатоспленомегалия, являются важными факторами для определения необходимости гемотрансфузии.
Классические симптомы тяжелой формы заболевания включают талассемические фации (гиперплазия верхней челюсти, плоская переносица, выступающие лобные бугры), патологические переломы костей, выраженную гепатоспленомегалию и кахексию и в основном наблюдаются в странах, в которых нет возможностей для проведения длительной гемотрансфузионной терапии. Иногда у пациентов с умеренной анемией эти особенности развиваются вследствие выраженного неэффективного компенсаторного эритропоэза.
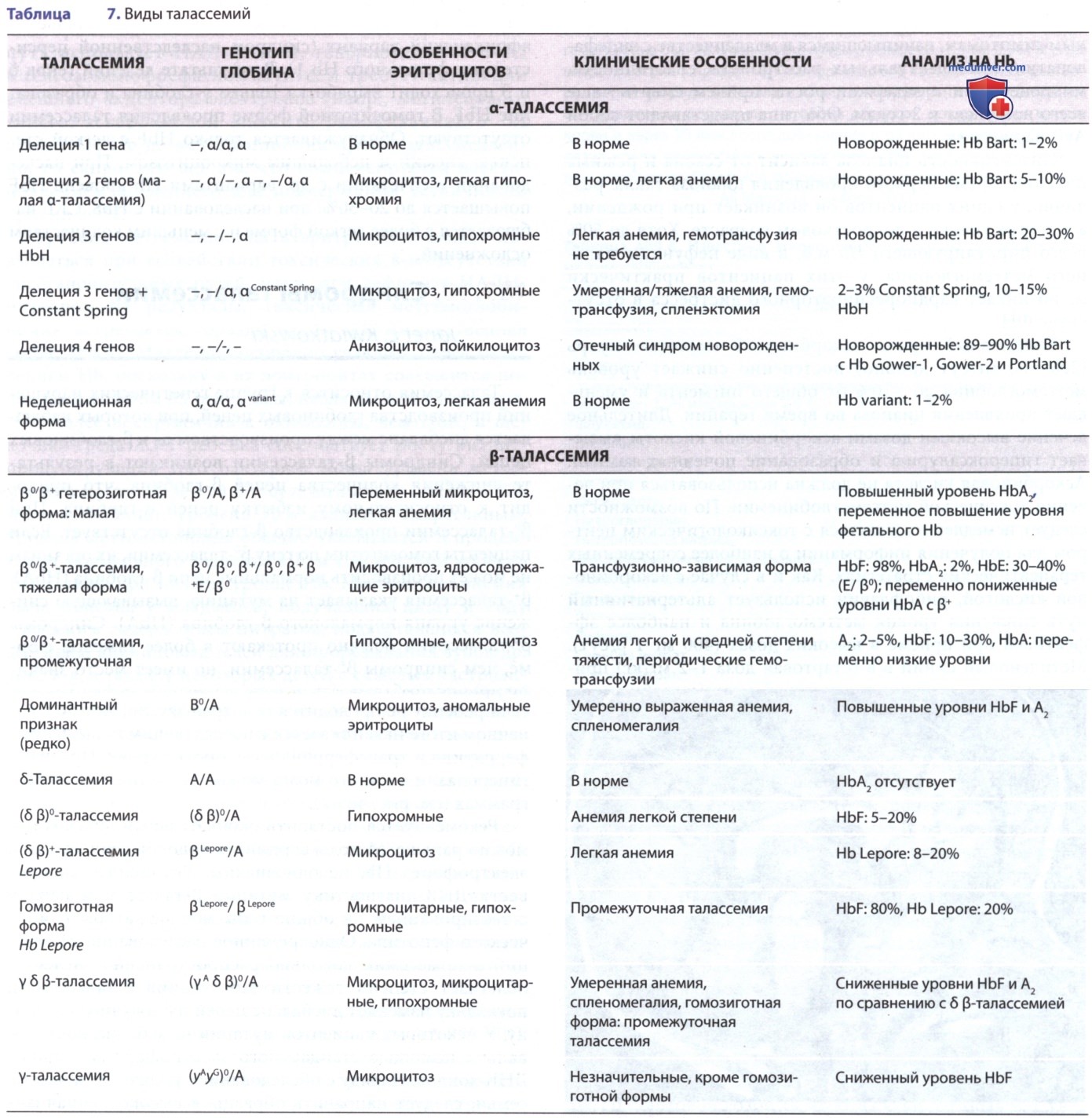
У пациентов с неэффективным эритропоэзом, которым не была проведена гемотрансфузия, может развиться выраженная спленомегалия с гиперспленизмом и абдоминальными симптомами. К особенностям неэффективного эритропоэза относятся расширенные костномозговые полости (со значительным расширением костного мозга в области лица и черепа), экстрамедуллярный гемопоэз и более высокие метаболические потребности (рис. 1). Хроническая анемия и повышенная эритроидная активность приводят к увеличению всасывания железа из ЖКТ и вторичному повреждению органов при гемосидерозе.
Рисунок 1. Неэффективный эритропоэз у пациента 3 лет с большой β-талассемией, которому не проводилась гемотрансфузия: A — магнитно-резонансная томограмма обширного расширения костномозговых полостей черепа; B— обычное рентгенографическое изображение трабекул; C — компьютерная томограмма изображение облитерации верхнечелюстных пазух кроветворной тканью
Длительная гемотрансфузионная терапия резко улучшает качество жизни и уменьшает осложнения при талассемии тяжелой степени. Гемосидероз, обусловленный гемотрансфузией, становится основным клиническим осложнением трансфузионно-зависимой талассемии. Каждый 1 мл эритроцитарной массы содержит ~1 мг железа. Не существует физиол. механизма для устранения избытка железа в организме. Железо сначала откладывается в печени, а затем в эндокринных органах и сердце. Это приводит к интенсивному развитию гипотиреоза, гипогонадотропного гонадизма, дефицита гормона роста, гипопаратиреоза и СД. Отложение железа в сердце вызывает СН и аритмию, а болезни сердца являются основной причиной смерти у плохо хелатированных пациентов.
В конечном счете большинство пациентов, не получающих соответствующую железо-хелатирующую терапию, умирают от СН/аритмии вследствие гемосидероза. Заболеваемость, индуцированная накоплением железа, м.б. предотвращена проведением соответствующей железо-хелатирующей терапии.
2. Результаты лабораторных исследований. В США некоторым детям с большой β-талассемией диагноз ставится при проведении скрининга новорожденных в результате обнаружения только фетального Hb при электрофорезе. Тем не менее при выявлении во время скрининга у новорожденных с мутациями β+ некоторого количества HbА эти дети выпадают из поля зрения врачей. Паттерн HbFE м.б. сопоставим с β0-талассемией HbЕ/более доброкачественным заболеванием HbЕЕ и должен быть под наблюдением. Из-за того, что не существует стандартизированной неонатальной диагностики талассемии, необходимо тщательно наблюдать за новорожденными с неясными талассемическими мутациями и детьми из этнических групп высокого риска.
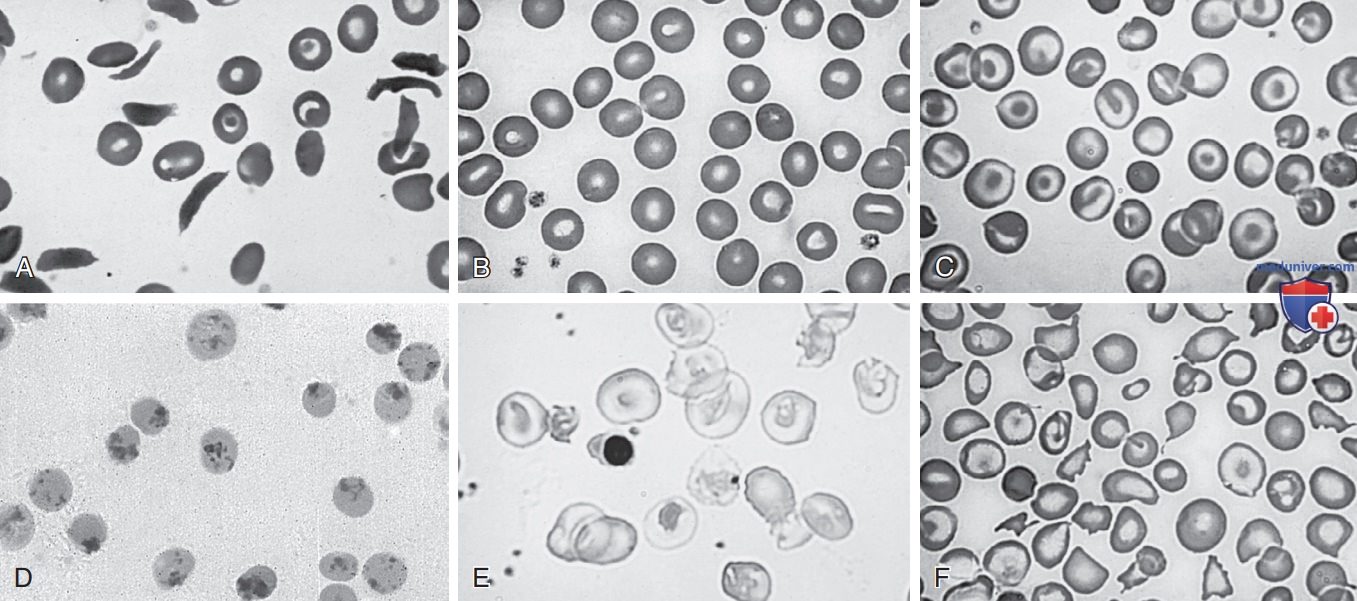
У младенцев с тяжелыми формами β-талассемии наблюдается прогрессирующая анемия после периода новорожденности. Эритроциты характеризуются микроцитозом, гипохромией и таргетированием. Обычно наблюдаются ядросодержащие эритроциты, выраженный анизопойкилоцитоз и относительная ретикулоцитопения (см. рис. 2). Если не проводится гемотрансфузия, уровень Hb неуклонно падает, часто до <6 г/дл.
Рисунок 2. Морфология эритроцитов при гемоглобинопатиях: A — серповидно-клеточная анемия — мишеневидные клетки и фиксированные (необратимо серповидные) клетки; B — серповидно-клеточная аномалия эритроцитов (гемоглобин AS) — нормальная морфология эритроцитов; C — гемоглобин СС — мишеневидные клетки и единичные сфероциты; D — врожденная анемия с тельцами Гейнца (Heinz) (нестабильный гемоглобин) — эритроциты, окрашенные суправитальным красителем (бриллиантовым крезиловым синим), содержат в/клеточные включения; E — гомозиготная β0-талассемия — тяжелая гипохромия с деформированными эритроцитами и нормобластами; F — болезнь гемоглобина Н (α-талассемия) — анизопойкилоцитоз с мишеневидными клетками
Количество ретикулоцитов обычно составляет <8% и является неадекватно низким по сравнению со степенью анемии, вызванной неэффективным эритропоэзом. Уровень неконъюгированного сывороточного билирубина обычно повышен, но др. биохим. показатели изначально м.б. в норме.
Даже если ребенку не проводится гемотрансфузия, железо в конечном итоге накапливается с последующим повышением ферритина и трансферрина в сыворотке крови. Признаки гиперплазии костного мозга можно увидеть на рентгенограммах (см. рис. 1).
Рекомендуется поставить окончательный диагноз как можно раньше. Методы скрининга новорожденных, напр. электрофорез Hb, не однозначны. Рекомендуется провести ДНК-диагностику мутаций β-талассемии наряду с тестированием на общие гены-модификаторы клинического фенотипа. Одновременное наследование >1 делеций α-талассемии довольно распространенное явление, облегчает степень тяжести заболевания β-талассемии, поскольку изменяет дисбаланс цепей αβ в лучшую сторону. У некоторых пациентов мутации не м.б. диагностированы с помощью стандартного электрофореза/обычных ДНК-зондов. Наряду с обследованием родителей и членов семьи, следует направить образцы в высокоспециализированную лабораторию третьего уровня. После установления окончательного диагноза семьи должны пройти детальное консультирование.
3. Ведение и лечение талассемии:
- Гемотрансфузионная терапия. Большая β-талассемия — клинический диагноз, который требует интеграции лабораторных данных и клинических признаков. Из числа пациентов с гомозиготной β0-талассемией (наиболее тяжелыми мутациями) 15-20% могут иметь клиническое течение, по фенотипу соответствующее промежуточной талассемии.
Вместе с тем 25% пациентов с гомозиготной β+-талассемией, как правило, более доброкачественного генотипа, могут иметь трансфузионно-зависимую талассемию. Преходящие клинические явления, такие как внезапное падение уровня Hb на фоне инфицирования парвовирусом, требующее гемотрансфузии, не обязательно означают трансфузионную зависимость пациента. Для определения необходимости в длительной гемотрансфузионной терапии необходимо длительное наблюдение за клиническими проявлениями, напр. рост, костные изменения и уровень Hb.
- Рекомендации по гемотрансфузионной терапии. У пациентов, нуждающихся в гемотрансфузионной терапии, должен быть определен расширенный фенотип и/или генотип эритроцитов. Пациенты должны получать эритроциты, очищенные от лейкоцитов и, как минимум, соответствующие АГн D, С, с, Е, е и Kell. При трансплантации стволовых клеток те ложны быть безопасны с точки зрения инфицирования ЦМВ. Гемотрансфузии обычно следует проводить с интервалом 3-4 нед, с целью поддержания предтрансфузионного уровня Hb 9,5-10,5 г/дл. Необходим постоянный мониторинг инфекций, передающихся при гемотрансфузиях (HAV, HBV, HCV, ВИЧ), аллоиммунизации, ежегодных показаний к проведению переливаний крови и трансфузионных реакций.
- Мониторинг перегрузки железом. Избыточные запасы железа при проведении гемотрансфузий вызывают много осложнений при большой β-талассемии. Для оптимальной терапии необходима точная оценка избыточных запасов железа. Серия тестов на уровни ферритина сыворотки крови представляет собой эффективный скрининговый метод, позволяющий определить тенденции баланса железа в организме, но результаты не могут предсказать точное его количество. При ведении пациента только на основе показателей сывороточного ферритина может иметь место как недостаточное, так и чрезмерное лечение. Количественное измерение железа в печени и сердце по МРТ является стандартным неинвазивным методом измерения гемосидероза тканей; методы определения уровня отложений железа в ПЖЖ и половых органах находятся в стадии изучения.
Эта технология, наряду с многочисленными хелатирующими ЛП, позволяет проводить целенаправленную терапию пациентам с органоспецифическим гемосидерозом до начала проявлений функциональной недостаточности органов. Интеграция таких технологий визуализации с применением хелатирующей терапией может предотвратить развитие СН, СД и др. дисфункции органов.
Определение количественного содержания железа в печени с помощью утвержденных методов R2/R2* МРТ является лучшим способом выявления общих запасов железа в организме и должно проводится пациентам после начала длительной гемотрансфузионной терапии. Показатели содержания железа в печени помогут определить режим хелатирования. Для определения количественного содержание железа в сердце проводят Т2* МРТ ССС, обычно начиная с 10 лет, но при тяжелой перегрузке железом/если нет данных о ранее проведенных гемотрансфузиях и хелатирующей терапии, проводят раньше. Возможно, существует несоответствие между уровнями железа в печени и в сердце из-за разл. скорости загрузки и разгрузки тканей и неравномерного влияния хелатирующих ЛП на выведение железа из разных органов.
- Хелатирующая терапия. Железо-хелатирующая терапия должна назначаться, как только гемосидероз становится значительным. Как правило, такой момент наступает через год от начала гемотрансфузионной терапии и коррелирует с уровнем сывороточного ферритина >1000 нг/мл и/или уровнем концентрации железа в печени >5000 мкг/г в пересчете на массу сухого в-ва. В настоящее время в инструкции по применению железо-хелатирующих ЛП нет информации об использовании у детей <2 лет.
На данный момент есть 3 хелатора железа (дефероксамин, деферазирокс и деферипррн); каждый из них отличается по способу введения, фармакокинетике, побочным эффектам и эффективности. Комбинированная хелатирующая терапия может потребоваться при высокой нагрузке железом. Конечная цель состоит в предотвращении повреждения тканей, вызванного гемосидерозом, с попытками избежать токсичности хелатных соединений. Поэтому требуется тщательное наблюдение за пациентами. В целом токсичность хелатных соединений возрастает по мере уменьшения запасов железа.
Дефероксамин ("Десферал") является наиболее изученным хелатором железа; он имеет отличный профиль безопасности и эффективности. Этот ЛП требует п/к/в/в-введения вследствие его плохой биодоступности при приеме внутрь и короткого периода полувыведения <30 мин, что требует введения в виде непрерывной инфузии в течение не <8 ч ежедневно, 5-7 сут/нед. Дефероксамин назначается с 25 мг/кг, затем доза м.б. увеличена до 60 мг/кг пациентам с тяжелой перегрузкой железом. Основная проблема при использовании дефероксамина — недобросовестное соблюдение режима лечения из-за сложного пути введения, отнимающего массу времени. Неблагоприятные побочные эффекты включают местные кожные реакции, ототоксичность, изменения на сетчатке и дисплазию костной ткани с укорочением трубчатых костей.
В продаже в США также имеется хелатор железа для приема внутрь деферазирокс («Эксиджад», «Джадену»). 70% пациентов, получавших дефероксамин, перешли на деферазирокс из-за способа введения. Период полувыведения деферазирокса >16 ч, поэтому ЛП требует однократного ежедневного приема. В наличии имеются две формы ЛП: диспергируемая таблетка, растворяемая в воде/соке, и таблетка, покрытая пленочной оболочкой. Недавно FDA утвердило дозированную форму в виде гранул, которые добавляются в мягкую пищу с последующим употреблением внутрь. Разные составы деферазирокса назначаются в разных дозах. Для диспергируемой таблетированной формы («Эксиджад») стартовая доза обычно 20 мг/кг в сутки и м.б. увеличена до 40 мг/кг в сутки в зависимости от степени перегрузки железом.
Дозировка для таблеток, покрытых пленочной оболочкой, и гранул ("Джадену") на 30% ниже, чем для диспергируемых таблеток, со стартовой дозой 14 мг/кг в сутки, которая м.б. увеличена до максСД 28 мг/кг. Наиболее распространенными побочными эффектами являются симптомы со стороны ЖКТ, которые м.б. нивелированы назначением таблеток, покрытых пленочной оболочкой, поскольку они не содержат лактозы и натрия, содержащихся в диспергируемой таблетированной форме, которые, возможно, вызывают некоторые симптомы со стороны ЖКТ. Наиболее серьезным побочным эффектом деферазирокса является возможное поражение почек. До 30% пациентов имеют преходящее повышение уровня креатинина, что может потребовать временных изменений дозировки. Такая токсичность чаще возникает в условиях обезвоживания.
Длительные исследования с участием тысяч пациентов не выявили прогрессирующей почечной дисфункции, но отмечались единичные случаи почечной недостаточности. Кроме того, у ~8% пациентов может возникнуть печеночный трансаминит с превышением верхней границы нормы в >5 раз. Всем пациентам необходимо ежемесячно проводить развернутый биохим. анализ крови, а мониторинг на протеинурии должен быть постоянен.
Деферипрон ("Феррипрокс"), хелатор железа для приема внутрь, одобрен в США для применения в качестве ЛП второго ряда. Он имеет период полувыведения ~3 ч и требует назначения 3 р/сут. Стартовая доза 75 мг/кг в сутки м.б. увеличена до 99 мг/кг в сутки в зависимости от степени перегрузки железом. Деферипрон, представляющий собой небольшие молекулы, легко проникает в сердечную ткань и м.б. более эффективным, чем др. хелаторы, для уменьшения сердечного гемосидероза. Наиболее серьезным побочным эффектом деферипрона является преходящий агранулоцитоз, который возникает у 1% пациентов обычно на первом году лечения. Агранулоцитоз вызывал редкие случаи смерти, если пациенты не находились под соответствующим наблюдением.
Применение деферипррна требует частого контроля показателей крови, как правило, еженедельно в течение, по крайней мере, одного года терапии. Самое главное, при всех лихорадочных заболеваниях следует продолжать прием ЛП и определять уровень нейтрофилов.
Поскольку сейчас пациенты с талассемией живут дольше, цели хелатирования железа изменились. Агрессивное лечение с помощью комбинированной хелатирующей терапии часто назначается пациентам с тяжелой перегрузкой железом для предотвращения/обращения вспять дисфункции органов. Дефероксамин в комбинации с деферипроном обычно применяют у пациентов с повышенным содержанием железа в сердечной ткани. Комбинированная терапия (дефероксамином + деферазироксом)/(деферазироксом + деферипроном) также м.б. эффективной у пациентов с тяжелой перегрузкой железом.
- Гидроксимочевина. Гидроксимочевина, антиметаболит ДНК, увеличивает производство фетального Hb. Наиболее успешно она применялась при СКВ и у некоторых пациентов с промежуточной β-талассемией. Исследования в области большой β-талассемии малочисленны. Пациентам с промежуточной формой заболевания во многих странах мира назначается терапия гидроксимочевиной. Несмотря на повышение уровня фетального Hb, оно не имеет прогностической корреляции с увеличением уровня общего НЬ у этих пациентов. В целом, по-видимому, наблюдается среднее увеличение уровня НЬ на 1 г/дл (в диапазоне 0,1-2,5 г/дл).
Терапия промежуточной талассемии гидроксимочевиной снижает риск развития язв на ногах, легочной гипертензии и экстрамедуллярного гемопоэза. Стартовая доза при промежуточной талассемии 10 мг/кг и м.б. увеличена до 20 мг/кг в сутки. Пациенты с β-талассемией подвергаются повышенному риску развития цитопении при применении гидроксимочевины, вследствие чего, возможно, дозу ЛП повышать не следует. Необходим тщательный мониторинг ОАК с подсчетом лейкоцитарной формулы.
- Трансплантация гемопоэтических стволовых клеток. ТГСК помогла вылечить >3000 пациентов с большой β-талассемией. Среди пациентов из группы низкого риска с пересадкой от HLA-совместимых братьев/сестер наблюдается выживаемость, по крайней мере, 90%, а выживаемость без рецидива — 80%.
Обычно для предотвращения отторжения трансплантата и рецидива талассемии используются схемы миелоаблативного кондиционирования. Наиболее эффективным этот метод лечения показал себя у детей <14 лет без гемосидероза и гепатомегалии, перенесших аллогенную трансплантацию от HLA-совместимых братьев/сестер. Всем детям, у которых есть HLA-совместимый брат/сестра, должна быть предложена возможность трансплантации костного мозга. Альтернативные схемы трансплантации для пациентов без соответствующих доноров являются экспериментальными и не всегда эффективны. Методы генной терапии находятся в стадии изучения, предварительные результаты с использованием лентивек-торов были многообещающими, особенно у пациентов с генотипами β+/HbE β-талассемии.
- Спленэктомия. Спленэктомия может потребоваться пациентам с талассемией при гиперспленизме. У этих пациентов отмечены снижение стабильного уровня Hb и/или повышенная потребность в гемотрансфузии. Однако спленэктомия все реже используется в качестве варианта терапии; кроме риска инфицирования все чаще принимаются во внимание и др. серьезные побочные эффекты. При промежуточной талассемии пациенты с удаленной селезенкой имеют выраженное повышение риска венозного тромбоза, легочной гипертензии, язв ног и бессимптомного инфаркта ГМ по сравнению с пациентами без спленэктомии. Все пациенты должны быть полностью иммунизированы против инкапсулированных бактерий и получить соответствующие инструкции по лечению лихорадочных заболеваний.
После спленэктомии следует проводить пенициллинопрофилактику для предотвращения развития сепсиса, а семьи пациентов должны быть проинформированы о риске его развития и развития лихорадки.
4. Профилактический мониторинг пациентов с талассемией:
- Заболевания сердца. ССЗ являются основной причиной смерти при талассемии. Для оценки функции сердца и давления в ЛА следует проводить мониторинг методом серийной ЭхоКГ. Легочная гипертензия часто возникает у пациентов с талассемией, не зависящих от гемотрансфузий, и м.б. показанием к трансфузионной терапии. Через ~8 лет длительной гемотрансфузионной терапии может развиться гемосидероз сердца, поэтому рекомендуется проведение обследование методом Т2* МРТ. Пациенты с сердечным гемосидерозом и снижением ФВ нуждаются в интенсивной комбинированной хелатирующей терапии. Также >10 лет периодически проводится ЭКГ вследствие риска развития аритмии, обусловленной перегрузкой сердца железом.
- Эндокринные заболевания. Эндокринная функция постепенно снижается с возрастом вследствие гемосидероза и дефицита питательных в-в. Отложение железа в гипофизе и эндокринных органах может привести к множественным эндокринопатиям, включая гипотиреоз, дефицит гормона роста, задержку полового созревания, гипопаратиреоз, СД, остеопороз и недостаточность надпочечников. Мониторинг эндокринной дисфункции начинается рано, в ~5 лет/по крайней мере, после 3 лет длительной гемотрансфузии. Каждые 6 мес все дети нуждаются в измерении роста, МТ, оценке развития в пубертатном периоде и измерении роста в положение сидя.
Сканирование плотности костной ткани следует проводить, начиная со второго десятилетия жизни, учитывая высокую частоту развития остеопении. Необходима оценка содержания в организме питательных в-в.
Большинство пациентов нуждаются в восполнении витаминов D и С, цинка. Все большее внимание привлекает проблема фертильности пациентов, в связи с чем должно проводиться регулярное обследование.
- Психосоциальная поддержка. Талассемия создает серьезные проблемы в жизни семьи пациента и значительные препятствия для нормального развития ребенка. Необходимо заблаговременно проводить консультирование семей с учетом местной культурной специфики, так как пользование услугами социальных служб по уходу за детьми, начиная с ранних этапов заболевания, уменьшает психологическую травму, вызванную терапией. Обязательным является раннее консультирование социальными службами семей в отношении финансовых и социальных вопросов.
г) Другие синдромы β-талассемии:
1. Трансфузионно-независимая талассемия: промежуточная β-талассемия. Синдромы β-талассемии характеризуются снижением выработки β-глобиновых цепей HbА. Было описано 200-300 мутаций β-талассемии. Эти мутации могут влиять на любой этап транскрипции генов β-глобина. Как уже говорилось, при β0-талассемии отсутствует производство нормальных β-цепей, а производство HbА при β+-мутациях снижается. Некоторые мутации, напр. HbЕ, имеют структурные мутации хромосом. Др., напр. δβ-талассемия/наследственная персистенция фетального Hb, являются вариантами β-талассемии со снижением производства гена β-глобина и повышенной компенсаторной выработкой фетального Hb.
Поскольку корреляция фенотипа с генотипом варьирует, пациенты с β-талассемией в значительной степени классифицируются по клинической картине. Трансфузионно-зависимая талассемия/болыпая талассемия представляет собой группу наиболее тяжелых нарушений. Трансфузионно-независимая талассемия (промежуточная форма) включает целый спектр пациентов, которым в младенчестве изначально не требуется проведение длительной гемотрансфузионной терапии, но время от времени на протяжении всей жизни м.б. показания к ее назначению. Основной определяющей характеристикой таких пациентов является меньший дисбаланс α-β-глобиновой цепи, чем при большой талассемии. Иногда генетические модификаторы изменяют тяжесть первичной мутации и уменьшают дисбаланс глобиновой цепи.
Сочетанное наследование признака α-талассемии/полиморфизмов промоторов глобина, таких как BCL11, может уменьшить тяжесть заболевания и привести к развитию трансфузионно-независимой талассемии. HbЕ β-талассемия может иметь как трансфузионно-зависимую, так и трансфузионно-независимую форму. Эти вторичные генетические модификаторы играют определенную роль в изменении тяжести данного расстройства. Иногда пациенты с единственной мутацией β-талассемии/АуД малой β-талассемией имеют клинические признаки промежуточной/трансфузионно-независимой талассемии. Генетические исследования этих пациентов часто выявляют сочетанное наследование генетических модификаторов, ухудшающих состояние, таких как трипликация а-гена/нестабильная мутация β-глобина.
Пациенты с промежуточной талассемией имеют выраженный неэффективный эритропоэз, что приводит к микроцитарной анемии с уровнем Hb ~7 г/дл (6-10 г/ дл). Такие пациенты имеют некоторые осложнения, характерные для нетрансфузионной большой талассемии, но тяжесть их варьирует в зависимости от степени неэффективности эритропоэза. У них может развиваться медуллярная гиперплазия, гепатоспленомегалия, гемопоэтические псевдоопухоли, легочная гипертензия, язвы ног, тромботические явления и отставание в росте. У многих пациентов развивается гемосидероз вследствие повышенной абсорбции железа в ЖКТ, что требует проведения хелатирующей терапии. Экстрамедуллярный гемопоэз может возникать в позвоночном канале, сдавливая спинной мозг и вызывая неврологические симптомы; последнее является неотложным состоянием, требующим немедленной местной лучевой терапии для остановки эритропоэза.
Пациентам с промежуточной талассемией при тяжелых клинических проявлениях показана гемотрансфузионная терапия.
Малая талассемия часто ошибочно диагностируется как дефицит железа у детей, поскольку эти 2 диагноза приводят к сходным гематологическим аномалиям, выявляемым по результатам ОАК. Однако дефицит железа встречается гораздо чаще. Для выявления детей, нуждающихся в дальнейшем обследовании достаточно провести короткий курс железосодержащими ЛП с последующим повторным обследованием. Дети с малой β-талассемией имеют устойчиво нормальные показатели ширины распределения эритроцитов по объему и низкий показатель среднего объема эритроцитов, в то время как у пациентов с дефицитом железа при лечении повышаются показатели ширины распределения эритроцитов по объему. В анализе на Hb у пациентов с малой β-талассемией повышен уровень HbА2 и непостоянно повышен уровень HbF.
Существуют бессимптомные формы малой β-талассемии, и, если семейный анамнез вызывает подозрение, м.б. показано дальнейшее обследование.
д) Синдромы α-талассемии. То же давление эволюционного отбора, вызвавшее β-талассемию и СКБ, вызвало и а-талассемию. Младенцы с α-талассемией выявляются в период новорожденности по повышенному уровню синтеза Hb Bart (γ4) во время фетального периода и по его наличию при рождении. Синдромы α-талассемии наиболее часто встречаются в Юго-Восточной Азии. При данном заболевании наиболее часто обнаруживаются делеционные мутации. В дополнение к ним существуют неделеционные мутации гена α-глобина, наиболее распространенной из которых является Constant Spring (αCSα); эти мутации вызывают более тяжелые анемию и клиническое течение, чем делеционные мутации. В норме существует 4 гена α-глобина.
Разл. фенотипы при α-талассемии в значительной степени обусловлены тем, происходит ли делеция одного (α+-талассемия)/обоих (α0-талассемия) генов α-глобина в каждом из 2 локусов.
Делеция одного гена α-глобина (бессимптомный признак) не определяется гематологически. В частности, не наблюдается изменений в показателях ширины распределения эритроцитов по объему и среднего содержания НЬ в эритроцитах. Пациенты с этой делецией обычно выявляются после рождения с делецией 2 гена/HbН (04), но при проведении некоторых программ скрининга новорожденных были зарегистрированы даже низкие концентрации Hb Bart. В период новорожденности наблюдается <3% Hb Bart. Делеция одного гена α-глобина часто встречается у афроамериканцев.
Делеция двух генов α-глобина приводит к развитию малой α-талассемии. Аллели α-глобина м.б. утрачены в конфигурации транс (-α/-α)/цис (α,α/-SEA). Транс-/цис-мутации могут сочетаться с др. мутациями/делециями и приводить к формированию НЬН/развитию большой α-талассемии. У лиц из Африки/африканского происхождения наиболее распространенные делеции α-глобина находятся в транс-конфигурации, тогда как у лиц из Азии/средиземноморского региона/их потомков наиболее распространены цис-делеции.
Малая α-талассемия (два отсутствующих гена α-глобина) проявляется в виде микроцитарной анемии, которую можно ошибочно принять за ЖДА (см. рис. 2). Анализ на НЬ в норме, за исключением периода новорожденности, когда Hb Bart обычно составляет <8%, но >3%. Клинические проявления делеции 2 генов α-глобина обычно ошибочно расцениваются как железодефицитные, учитывая наличие низких показателей как ширины распределения эритроцитов по объему, так и среднего содержания Hb в эритроцитах. Самый простой подход к ДД дефицита железа и малой α-талассемии — тщательное изучение анамнеза питания. У детей с ЖДА часто в пищевом рационе не хватает железа, и вместе с этим они употребляют большое количество коровьего молока.
Как вариант, краткий курс приема железосодержащих ЛП наряду с мониторингом параметров эритроцитов может подтвердить этот диагноз.
Если оба родителя ребенка, у которого диагностирована малая а-талассемия, являются носителями цис-конформации, то они находятся в группе риска по развитию отечного синдрома новорожденных. Соответственно следует проводить семейный скрининг и генетическое консультирование.
Делеция 3 генов α-глобина подразумевает диагноз болезни HbН. Более тяжелая форма этого заболевания м.б. вызвана неделеционной мутацией а-глобина в сочетании с делециями 2 генов. HbН Constant Spring (-α/α, αCS) является наиболее распространенным типом неделеционной болезни HbН.
В Калифорнии, где проживает большое количество лиц азиатского происхождения, распространенность болезни HbН 1:10 000 всех новорожденных. Самый простой способ диагностики данной патологии — обследование в период новорожденности, когда наблюдается избыток γ-тетрамеров, а Hb Bart обычно составляет >25%. Полезно подтверждение диагноза у родителей. В более позднем детском возрасте наблюдается избыток тетрамеров β-глобиновой цепи, что приводит к образованию HbН. Окончательный диагноз требует анализа ДНК. Краситель бриллианткрезил блау может окрашивать HbН, но он редко используется для диагностики.
Пациенты с болезнью HbН имеют выраженный микроцитоз, анемию, легкую спленомегалию и, иногда, иктеричность склер и ЖКБ. Длительная гемотрансфузионная терапия обычно не требуется, поскольку уровень Hb находится в диапазоне 7-11 г/дл, при ширине распределения эритроцитов по объему 51-73 фл, но могут потребоваться нерегулярные гемотрансфузии при усилении анемии. Пациенты с неделеционной болезнью HbН чаще нуждаются в гемотрансфузиях, чем пациенты с делеционной формой.
Делеция всех 4 аллелей гена α-глобина вызывает тяжелую анемию в течение фетального периода, что приводит к развитию отечного синдрома новорожденных; для выживания плод должен иметься в наличии ген (-глобина. При рождении нормальных Hb у ребенка нет (в первую очередь Hb Bart с Hb Gower-1, Gower-2 и Portland). В/маточные гемотрансфузии могут спасти плод, но часто этот метод приводит к развитию врожденных аномалий и задержке развития НС. Младенцы с тяжелой формой а-талассемии будут зависимы от гемотрансфузий на протяжении всей жизни, и ТГСК является единственным способом излечения.
Лечение болезни HbН требует постоянного мониторинга роста и дисфункции органов. Показано обогащение пищевого рациона ЛП с фолиевой кислотой и поливитаминами без железа. У пациентов старшего возраста может уменьшиться плотность костной ткани при дефиците кальция и витамина D. Для укрепления костей показаны витамина D при низком его уровне и адекватное потребление кальция с пищей. Следует избегать приема железосодержащих ЛП, так как пациенты подвержены риску развития гемосидероза. Периодически может возникать необходимость проведения гемотрансфузии во время интеркуррентной инфекции, особенно при неделеционной форме болезни HbН.
Иногда показана спленэктомия, и вследствие высокого риска постспленэктомического тромбоза следует рассмотреть возможность назначения ацетилсалициловой кислоты («Аспирина»)/др. АКТ после операции. Гемосидероз вследствие абсорбции железа в ЖКТ/гемотрансфузионного воздействия может развиться у пациентов старшего возраста, им может потребоваться хелатирующая терапия. Поскольку HbН является нестабильным Hb, чувствительным к окислительному повреждению, следует избегать оксидативных ЛП. Должны быть выявлены семейные пары из группы риска по отечному синдрому новорожденных, им должна быть предложена молекулярная диагностика тканей плода на ранних сроках беременности. На более поздних сроках в/маточная гемотрансфузия может увеличить выживаемость, но выжившим детям потребуется длительная гемотрансфузионная терапия/трансплантация костного мозга.