а) Нарушения дифференцировки и поляризации энтероцитов. Эта группа в основном включает два заболевания, характеризующихся типичными гистологическими и ультраструктурными нарушениями в биоптатах кишечника: БВМ и врожденная тафтинговая энтеропатия. Трихо-гепато-энтеральный синдром или синдромальная (фенотипическая) диарея также обычно классифицируются в этой группе.
б) Болезнь включений микроворсинок (врожденная атрофия микроворсинок). БВМ — АуР-расстройство, которое манифестирует при рождении обильной водянистой секреторной диареей. Также был описан вариант с поздним дебютом — через 2-3 мес после родов. Это самая серьезная причина врожденной диареи, связанной с развитием слизистой оболочки кишечника. Световая микроскопия слизистой оболочки тонкой кишки выявляет диффузное истончение слизистой оболочки с гипопластической атрофией ворсинок и отсутствием воспалительного инфильтрата. Диагностика проводится с помощью световой микроскопии с использованием окрашивания на PAS (periodic acid-Shiff — фуксинсернистая кислота, реактив Шиффа) и CD 10, которое выявляет очень тонкую или отсутствующую щеточную кайму вместе с PAS-«+» и CD10-«+» в/клеточными включениями. Электронная микроскопия выявляет энтероциты с отсутствующими или редкими микроворсинками.
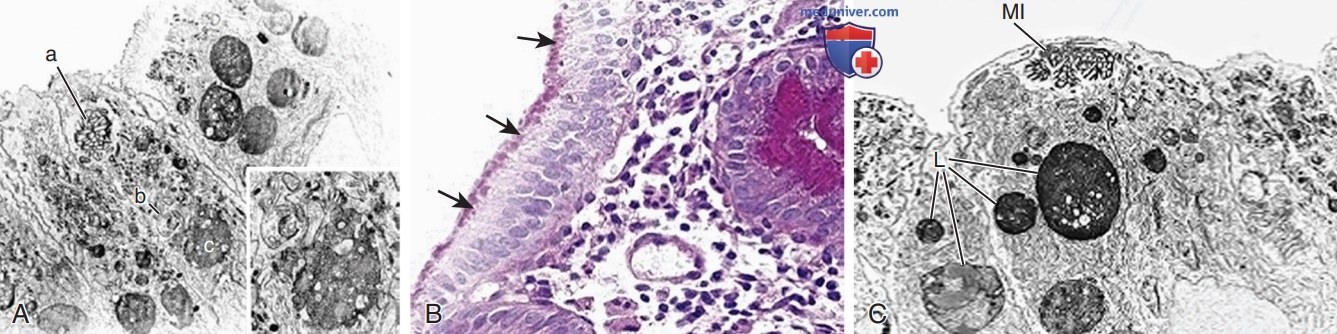
Апикальная цитоплазма энтероцитов содержит электронно-плотные секреторные гранулы; отличительным признаком является наличие микроворсинок в завернутой внутрь энтероцита апикальной мембране (рис. 1). При пренатальном УЗИ наблюдается многоводие, и у новорожденных обычно очень рано начинается тяжелая водянистая диарея (до 200-330 мл/кг в сутки), вызывающая обезвоживание и низкий темп физического развития. Несмотря на парентеральное питание, диарея продолжается, и при этом трудно контролировать водно-электролитный баланс. Сочетание БВМ и синдрома Фанкони, описанное у двух детей, могут осложнить ведение из-за дополнительных явлений ацидоза почечных канальцев, фосфатурии, рахита и потери жидкости почками.
Рисуснок 1. Болезнь включений микроворсинок: а — сверху вниз: микроворсинчатые включения (A), гранула с небольшим количеством микроворсинок (B) и лизосома (C), обнаруженная в том же энтероците. Вставка: Более высокое увеличение b и с х11 000, вставка х21 500; б — болезнь микро-ворсинчатых включений. Окрашивание кислым реактивом Шиффа позволяет выявить обилие «+» материала (стрелки) в апикальной части цитоплазмы энтероцитов; в — болезнь микроворсинчатых включений. Энтероцит на поверхности ворсины лишен микроворсинок с щеточной каймой, в то время как их апикальная цитоплазма содержит микроворсинчатое включение (Ml) и многочисленные лизосомы (L) х5500 А.
В когорте пациентов с ранним дебютом БВМ были обнаружены ассоциированные с этим заболеванием мутации гена MYO5B, кодирующего моторный белок, миозин Vb.
Мутации MYO5B приводят к дислокации апикальных белков и нарушению поляризации энтероцитов, что приводит к БВМ. Др. ген, t-SNARE синтаксин-3 (STX3), был описан у пациентов с более легким фенотипом БВМ. У пациентов с мутациями в связывающем белке STX3 STXBP2/Munc18-2, вызывающими семейный гемофагоцитарный лимфогистиоцитоз 5-го типа, также наблюдается атрофия микроворсинок и гистологические признаки, напоминающие БВМ. Потеря STX3 или Munc18-2 ингибирует слияние везикул с апикальной мембраной, что приводит к в/клеточной задержке апикальных белков. Мутации MY05B также были выявлены у нескольких пациентов с фенотипом, похожим на прогрессирующий семейный в/печеночный холестаз, но с нормальной активностью γ-глутамилтрансферазы в сыворотке крови и без патологии кишечника.
в) Тафтинговая энтеропатия (врожденная тафтинговая энтеропатия). Врожденная тафтинговая энтеропатия (дисплазия эпителия кишечника) манифестирует в первые недели жизни персистирующей водянистой диареей. Врожденная тафтинговая энтеропатия составляет небольшую долю младенцев с трудноизлечимой диареей. Отличительной особенностью гистологического строения слизистой оболочки тонкой кишки являются отдельно расположенные эпителиальные «пучки» («тафты» от англ. tufts) (каплевидные группы плотно упакованных энтероцитов с апикальным округлением плазматической мембраны), занимающие 80-90% поверхности эпителия. В типичных случаях патология не проявляется сразу после рождения. Тафты на поверхности эпителия могут наблюдаться и при других энтеропатиях.
Врожденная тафтинговая энтеропатия — фенотипически и генетически гетерогенное заболевание. Генетические исследования выявили мутации в гене ЕРСАМ у 73% пациентов и мутации в гене ингибитора активатора фактора роста гепатоцитов 2-го типа SPINT2/HAI2 в 21% случаев. Лишь у немногих пациентов не выявлено никаких явных мутаций. Фенотип, связанный с мутациями ЕРСАМ, обычно представляет собой изолированную врожденную диарею без сопутствующих дополнительных внекишечных симптомов, за исключением позднего дебюта артрита или поверхностного точечного кератита. При синдромной форме врожденной тафтинговой энтеропатии диарея связана с наличием одной или более из этих аномалий: поверхностный точечный кератит (100%), атрезия хоан (50%), атрезия пищевода или кишечника, анальная атрезия, дисплазия волос, поражение кожи, аномалии костей, гексадактилия и дисморфизм лица.
Специфического лечения не существует как для детей с БВМ, так и для пациентов с врожденной тафтинговой энтеропатией. Требуется постоянное полное парентеральное питание и при возможности выполнение трансплантации кишечника.
г) Трихо-гепато-энтеральный синдром (синдромальная диарея). Трихо-гепато-энтеральный синдром, также известный как синдромальная диарея, является врожденной энтеропатией, проявляющейся ранним дебютом тяжелой диареи. Пациенты рождаются маленькими для гестационного возраста. Диарея у них начинается в первые 6 мес жизни. Младенцы имеют аномальный фенотип, который характеризуется дисморфизмом лица (с выступающим лбом, широким носом и гипертелоризмом), с особой аномалией волос, узловатым трихорексисом. Волосы пушистые, легко удаляются и плохо пигментированы. Могут наблюдаться аномалии кожи, в т.ч. окраска «кофе с молоком» на нижних конечностях.
Поражение печени с развитием тяжелого фиброза или цирроза выявляют примерно у половины пациентов. Были отдельные сообщения о сердечных аномалиях и колите, а также об одном случае, связанном с многоводием, аномалиями плаценты и врожденным гемохроматозом. У пациентов могут быть дефектный гуморальный ответ, несмотря на нормальный уровень Ig в сыворотке крови, и дефектные АГн-специфичные кожные тесты, несмотря на «+» пролиферативные реакции in vitro. Трихо-гепато-энтеральный синдром также может трактоваться как ВЗК с очень ранним началом. При гистологическом исследовании можно обнаружить неспецифическую атрофию ворсинок с инфильтрацией собственной пластинки мононуклеарами или без нее и без специфических изменений эпителия. Мутации в гене 37-го домена тетратрикопептидного повтора (ТТС37) (60%) или SKIV2L (40%) были идентифицированы как причина трихо-гепато-энтерального синдрома.
Энтероциты с мутациями ТТС37 демонстрируют сниженную экспрессию натрий-протонных обменников (NHE)-2 и -3, ассоциированных с щеточной каймой аквапорина-7, Na+/I-симпортера и Н+/К+-АТФазы или неправильную их локализацию по сравнению с нормой. Прогноз этого вида трудноизлечимой диареи в младенчестве неблагоприятен. Долгосрочное наблюдение за этими детьми показало, что к 15 годам 50% пациентов были живы или им удалось полностью отменить полное парентеральное питание. Основными осложнениями являются патология печени и инфекции. Большинство детей имеют низкорослость, а половина слегка отстают в развитии.
1. Дефекты дифференцировки энтероэндокринных клеток. Этот класс врожденных диарей характеризуется аномальным развитием или функционированием энтероэндокринных клеток. Гены, вызывающие эти нарушения, кодируют либо факторы транскрипции, необходимые для развития всех или отдельных видов энтероэндокринных клеток, либо клеточные белки — эндопептидазы, которые необходимы для продукции активных гормонов из про-гормонов. Эти состояния проявляются осмотической диареей и в некоторых случаях дополнительными системными эндокринными нарушениями. Лечение заключается в нутритивной поддержке и гормональной заместительной терапии, если это необходимо. Были обнаружены четыре гена, связанные с заболеваниями, классифицированными в этой группе: NEUROG3, RFX6, ARX и PCSK1.
2. Кишечный анэндокриноз. NEUROG3 является ключевым фактором транскрипции, который контролирует судьбу эндокринных клеток как в ПЖЖ, так и в кишечнике. Мутации гена NEUROG3 вызывают тяжелую мальабсорбцию, рвоту, диарею, низкие темпы физического развития, обезвоживание и гиперхлоремический метаболический ацидоз. Пероральное питание чем-либо, кроме воды, вызывает диарею. Архитектоника ворсин и крипт в биоптатах тонкой кишки в норме, но окрашивание нейроэндокринных клеток (напр., с использованием АТл к хромогранину) демонстрирует полное отсутствие этой секреторной клеточной линии с сохранением бокаловидных клеток и клеток Панета.
3. Дефицит пропротеин-конвертазы 1/3. Передающийся АуР-дефицит пропротеин-конвертазы 1/3 (РС1/3), вызванный мутациями в гене PCSK1, характеризуется тяжелой врожденной диареей с мальабсорбцией, ранним ожирением и др. эндокринными аномалиями. Все регуляторные гормоны, вырабатываемые эндокринными клетками, в т. ч. в кишечнике, метаболизируются с помощью специфической Са2+-зависимой сериновой эндопротеазой, называемой пропротеиновой конвертазой 1/3 (также известной как нейроэндокринная конвертаза 1). Хроническая водянистая неонатальная диарея описана у младенцев с гиперинсулинизмом, гипогликемией, гипогонадизмом и гипоадренализмом. Биопсия тонкой кишки выявляет неспецифическую энтеропатию.
Обычно наблюдается дефицит СТГ, недостаточность надпочечников, несахарный диабет центрального типа и гипогонадизм.
д) Синдром Митчелла-Райли. Синдром Митчелла-Райли представляет собой сложный клинический фенотип, который включает тяжелое замедление в/утробного развития, неонатальный СД, аномалии ЖКТ (кольцевидная ПЖЖ, мальротация кишечника, агенезия желчного пузыря, аномалии ЖВП) и хроническую осмотическую диарею. Несколько пробандов детей с синдромом Митчелла-Райли, о которых ранее сообщалось, имели мутации RFX6. ДНК-связывающий белок RFX6 (регуляторный фактор Х6; кодируется RFX6) является фактором транскрипции крылатой спирали после сигнала нейрогенина-3, необходимого для развития островковых клеток и функции энтероэндокринных клеток. Иммунофлуоресцентное окрашивание биоптатов ПЖЖ мышей с заблокированным геном RFX6 показывает, что эндокринные клетки ПЖЖ присутствуют, но не экспрессируют гормоны островковых клеток, включая инсулин, глюкагон, соматостатин или грелин.
е) Мутации гена aristaless-related homebox. Ген ARX (aristaless-related homebox) кодирует гомеодомен, содержащий фактор транскрипции, необходимый для нормального развития энтероэндокринных клеток мыши и человека. Экспрессия ARX обнаруживается в субпопуляции нейрогенин-3-позитивных эндокринных предшественников, а также в субпопуляции гормон-продуцирующих клеток. У мышей удаление ARX из развивающейся эндодермы приводит к уменьшению некоторых типов энтероэндокринных клеток, таких как гастрин, глюкагон/GLP-l, холецистокинин, клеток, продуцирующих секретин и увеличение количества клеток, экспрессирующих соматостатин. Мутации в гене ARX связаны со сложным клиническим фенотипом: Х-сцепленной умственной отсталостью, судорогами, лиссэнцефалией, аномалией половых органов и иногда врожденной диареей.
ж) Аутоиммунная энтеропатия. Термин «аутоиммунная энтеропатия» описывает младенцев с тяжелой, затяжной диареей, отсутствием реакции на элиминационную диету, наличием циркулирующих аутоАТл к клеткам кишечника и/или сопутствующими аутоиммунными заболеваниями и отсутствием тяжелого иммунодефицита. Симптомы аутоиммунной энтеропатии обычно отмечаются после первых 6 мес жизни, проявляясь хронической диареей, энтеропатией с потерей белка, мальабсорбцией и низкими темпами физического развития. Диагноз основан на эндоскопическом и гистологическом выявлении воспаления главным образом в тонкой, но также и в толстой кишке. Гистологически в тонкой кишке обнаруживают частичную или полную атрофию ворсин, гиперплазию крипт и увеличение количества мононуклеаров в собственной пластинке.
У части пациентов может наблюдаться выраженный интраэпителиальный лимфоцитоз, напоминающий целиакию. Также могут выявляться криптиты и крипт-абсцессы, скрывающие наличие апоптоза. Иммунологические тесты позволяют обнаружить наличие аутоАТл, включая АТл против энтероцитов (присутствуют у 85% пациентов), а также к ассоцированному с аутоиммунной энтеропатией АГн 75 кДа.
ДД аутоиммунной энтеропатии у детей включает др. иммуноопосредованные расстройства, такие как энтеропатия, индуцированная белками коровьего молока, целиакия, болезнь Крона и реакция «трансплантат против хозяина». Важно исключить наличие первичного иммунодефицита, особенно у мальчиков с др. аутоиммунными заболеваниями, чтобы исключить IPEX-синдром. Описаны разл. фенотипы пациентов с IPEX-синдромом, а также IPEX-подобные формы аутоиммунной энтеропатии, которые не зависят от FOXP3 и встречаются у девочек с внекишечными аутоиммунными расстройствами или без них.
Возможности лечения ограничены и основаны на нутритивной поддержке, включая парентеральное питание, и назначении глюкокортикоидов с последующим назначением иммуносупрессивных ЛП. Трансплантация гемопоэтических стволовых клеток показана пациентам с известным молекулярным дефектом, таким как IPEX-синдром.
з) Аутоиммунный полигландулярный синдром 1-го типа:
1. Дефекты транспорта и метаболизма липидов. После поглощения из просвета жирные кислоты и моноацилглицерин транспортируются в эндоплазматический ретикулум. В эндоплазматическом ретикулуме они превращаются в триглицериды в ходе метаболических превращений, протекающих в несколько стадий, последняя из которых зависит от DGAT1. Аполипопротеин В (АроВ) и микросомальный белок переноса триглицеридов (МТТР) действуют согласованно, чтобы включать триглицериды в хиломикроны. Новообразованные хиломикроны формируются в эндоплазматическом ретикулуме в прехиломикронном транспортном пузырьке, который впоследствии сливается с комплексом Гольджи в процессе, зависимом от Sarlb. Затем хиломикрон транспортируется в пузырьке к базальной мембране, где он выходит из клетки.
и) Абеталипопротеинемия. Абеталипопротеинемия — синдром Бассена-Корнцвайга — редкое АуР-нарушение метаболизма липопротеинов, связанное с тяжелой мальабсорбцией жира/стеатореей с рождения. Уже на первом году жизни дети имеют низкие темпы физического развития, у них светлый дурно пахнущий и обильный стул. Живот вздут, а глубокие сухожильные рефлексы вследствие периферической нейропатии отсутствуют. Периферическая нейропатия вторична по отношению к дефициту витамина Е. Интеллектуальное развитие, как правило, замедлено. После 10 лет кишечные симптомы становятся менее выраженными, но может развиться атаксия с потерей ощущения положения собственного тела и потерей ощущения вибрации, а также развитием интенционного тремора. Эти симптомы отражают поражение проводящих путей задних рогов, мозжечка и базальных ганглиев. В подростковом возрасте при отсутствии введения адекватных доз витамина Е развивается атипичный пигментный ретинит.
Диагноз подтверждается наличием акантоцитов в мазке периферической крови и чрезвычайно низким уровнем холестерина в плазме (<50 мг/дл). Уровень триглицеридов также очень низкий (<20 мг/дл). Хиломикроны и липопротеины очень низкой плотности не обнаруживаются, а фракция ЛПНП практически отсутствует в кровотоке. Происходит выраженное накопление триглицеридов в энтероцитах ворсин слизистой ДПК. Пациенты с абеталипопротеинемией имеют мутации гена МТТР, который катализирует перенос триглицеридов к формирующимся частицам АроВ в эндоплазматическом ретикулуме.
Специфическое лечение отсутствует. Следует назначать нутритивную поддержку и большие дозы жирорастворимых витаминов A, D, Е и К. Витамин Е (100-200 мг/кг в сутки), по-видимому, останавливает прогрессирование неврологической симптоматики и дегенерацию сетчатки. Ограничение потребления длинноцепочечных жиров может облегчить кишечные симптомы. Для восполнения потребности в жире могут использоваться среднецепочечные триглицериды.
к) Гомозиготная гипобеталипопротеинемия. Гомозиготная гипобеталипопротеинемия является доминирующим наследственным заболеванием, связанным с мутациями в гене АРОВ, кодирующем АроВ, аполипопротеин зарождающегося хиломикрона. Гомозиготная форма неотличима от абеталипопротеинемии. Родители этих пациентов как гетерозиготы имеют сниженные концентрации ЛПНП и апопротеина В плазме крови, в то время как родители пациентов с абеталипопротеинемией имеют нормальные уровни этих показателей. При трансмиссионной электронной микроскопии биоптатов тонкой кишки размер липидных вакуолей в энтероцитах различается между абеталипопротеинемией и гипобеталипопротеинемией: при гипобеталипопротеинемии присутствует много мелких вакуолей, а при абеталипопротеинемии видны более крупные вакуоли.
л) Болезнь задержки хиломикронов (болезнь Андерсона). Болезнь задержки хиломикронов — редкое АуР-заболевание, вызванное мутациями в гене SAR1B. Мутации SAR1B приводят к нарушению перемещения зарождающихся хиломикронов в предхиломикронных транспортных везикулах между эндоплазматическим ретикулумом и аппаратом Гольджи, препятствуя успешной сборке хиломикронов и их доставке в собственную пластинку. У пациентов с болезнью задержки хиломикронов наблюдается стеаторея, хроническая диарея и низкие темпы физического развития. Акантоцитоз встречается редко, и неврологические проявления менее выражены, чем те, которые наблюдаются при абеталипопротеинемии. Уровень ХС в плазме умеренно снижен (<75 мг/дл), триглицериды натощак в норме, но уровни жирорастворимых витаминов, особенно А и Е, очень низкие. Лечение заключается в ранней агрессивной терапии жирорастворимыми витаминами и модификации потребления жиров, как при лечении абеталипопротеинемии.
м) Мутация диацилглицерин-1-ацилтрансферазы. DGAT1 кодирует диацил-КоА:ацилтрансферазу диацилглицерола (DGAT), которая преобразует диацилглицериды в триглицериды путем добавления ацилКоА. В тонкой кишке DGAT1 помогает собирать триглицериды, тогда как в печени он участвует в синтезе триглицеридов из жирных кислот, синтезированных de novo или поступающих из кровообращения. Механизм, посредством которого мутации DGAT1 вызывают диарею, неясен, но, вероятно, связан с накоплением липидных субстратов DGAT1 в энтероцитах или в просвете кишечника. Мутации в гене DGAT1 были зарегистрированы у пациентов с низким темпом физического развития, энтеропатией с потерей белка, гипоальбуминемией, диареей с ранним дебютом и рахитом, рефрактерным к приему витамина D внутрь.
н) Болезнь Вольмана. Болезнь Вольмана — редкое, смертельное заболевание, связанное с накоплением липидов во многих органах, включая тонкую кишку. В дополнение к рвоте, тяжелой диарее и гепатоспленомегалии у пациентов наблюдается стеаторея в результате непроходимости лимфатических сосудов. Низкий уровень свободного ХС, доступного для стероидогенеза в надпочечниках, приводит к недостаточности надпочечников. Отличительным признаком заболевания является характерная субкапсулярная кальцификация надпочечников. Дефицит лизосомной кислой липазы является основной причиной заболевания. Лизосомная кислая липаза — фермент лизосом, который гидролизует сложные эфиры ХС и триглицериды в эндолизосомах. Мутации, приводящие к потере функции в гене LIPA, связаны с разл. фенотипами.
Гомозиготные и сложные гетерозиготные мутации, приводящие к полному дефициту лизосомной кислой липазы, вызывают болезнь Вольмана. Мутации, связанные с остаточной активностью лизосомной кислой липазы, вызывают болезнь накопления эфиров ХС, менее тяжелое заболевание, имеющее разнообразные фенотипические проявления. Признаки заболевания, общие у младенцев, детей и взрослых, включают повышенный уровень аминотрансфераз в сыворотке крови, дислипидемию, гепатомегалию, фиброз печени и цирроз. Болезнь Вольмана уже в неонатальном периоде может также сопровождаться холестазом и тяжелым заболеванием печени в качестве ее основного проявления. Гемофагоцитарный лимфогистиоцитоз был выявлен у нескольких младенцев с болезнью Вольмана. Отличительной чертой заболевания является наличие кальцификации надпочечников, наблюдаемой при визуализации, и точный диагноз устанавливается генетически.
Опубликовано несколько случаев трансплантации гемопоэтических стволовых клеток с разными исходами. Для лечения пациентов одобрена рекомбинантная заместительная терапия ферментами при дефиците лизосомной кислой липазы. Это лечение позволило некоторым младенцам с болезнью Вольмана достичь относительно нормальных показателей роста и улучшить выживаемость. У детей старшего возраста и взрослых ферментная терапия корректировала дислипидемию и приводила к значительному улучшению показателей функции печени.
о) Болезнь Танжера. Свободный клеточный ХС мобилизуется вместе с фосфолипидами через экспортный насос АВСА1, что приводит к его переносу на внеклеточный акцептор АроА-1 и образованию ЛПВП. Мутации, приводящие к потере функции в генах АВСА1 у пациентов с болезнью Танжера, вызывают накопление ХС в кишечнике, селезенке, миндалинах, рецидивирующую невропатию, оранжево-коричневые пятна на толстой и подвздошной кишке и диарею в ассоциации со снижением уровня ХС в плазме (АроА-1 и А-П). При этом в плазме ЛПВП практически не обнаруживаются. Специфическая терапия болезни Танжера до сих пор не разработана.
п) Ситостеролемия. Ситостерин и другие стеролы секретируются обратно в просвет кишечника преимущественно через стериновый насос, представляющий собой парные полутранспортеры ABCG5IG8. Мутации транспортеров ABCG5 (стеролин-1) и ABCG8 (стеролин-2) приводят к нарушению выделения стерола и приводят к увеличению всасывания пищевых стеринов. Это расстройство ассоциировано с ксантомами сухожилий, развитием атеросклероза и гемолизом. Уровень фитостеролов в плазме (в основном ситостерина) обычно составляет >10 мг/дл.
р) Мальабсорбция желчных кислот. Желчные кислоты являются детергентными соединениями, секретируемыми и выводимыми из печени, и отвечают за эмульгацию пищевых жиров, способствуя их перевариванию и всасыванию. В процессе энтерогепатической циркуляции 95% желчных кислот реабсорбируются в терминальном отделе подвздошной кишки и транспортируются обратно в печень. Апикальный Na+-зависимый транспортер солей желчной кислоты (ASBT) или транспортер желчных кислот подвздошной кишки отвечает за активный обратный захват желчных кислот в терминальном отделе подвздошной кишки. Мутации в гене ASBT/SLC10A2 встречаются очень редко и ответственны за первичную мальабсорбцию желчных кислот, заболевание, связанное с врожденной диареей, стеатореей и снижением уровня ХС в плазме крови. Неабсорбированные желчные кислоты стимулируют экскрецию хлоридов в толстой кишке, что приводит к диарее.
Вторичная мальабсорбция желчных кислот может быть результатом патологии подвздошной кишки, напр. при болезни Крона, и после резекции подвздошной кишки. Диагноз мальабсорбции желчных кислот, как правило, основывается на снижении удержания 75-селен-гомохолевой кислоты таурина, меченного радиоактивной меткой (75SeHCAT), увеличении синтеза желчных кислот (уровень С4 в сыворотке) или увеличении потери желчных кислот с калом. В клинической практике диагноз часто основывается на эффекте секвестрантов желчных кислот (напр., холестирамин или колесевелам*), которые также являются предпочтительным методом лечения этого расстройства.
P.S. * Отсутствует на российском рынке.
Хроническая неонатальная диарея также была описана при АуР-церебротендинозном ксантоматозе, который вызван врожденным нарушением синтеза желчной кислоты вследствие дефицита 27-гидроксилазы. У этих детей наблюдается развитие катаракты в подростковом возрасте и задержка развития. Характерным проявлением заболевания является и неонатальный холестаз. Ксантомы сухожилий развиваются на втором и третьем десятилетиях жизни. Важно своевременно поставить диагноз, поскольку лечение эффективно при приеме хенодезоксихолевой кислоты внутрь.
с) Энтеропатия с потерей белка. ЭПБ — редкое заболевание, вызванное разл. кишечными и внекишечными расстройствами и характеризующееся чрезмерной потерей белков плазмы в кишечнике. Клиническая картина пациентов с ЭПБ различна и зависит от основной причины, но обычно включает отек и гипопротеинемию. До рассмотрения ЭПБ следует исключить нарушение синтеза белка (недостаточное питание, заболевания печени), потерю белка через др. органы (почки или кожу) или его перераспределение (септические состояния). Расстройства, вызывающие ЭПБ, можно разделить на расстройства, вызванные потерей белка из-за воспаленной или аномальной поверхности слизистой оболочки или из-за нарушений в лимфатической системе кишечника, таких как первичная или вторичная лимфангиоэктазия кишечника (табл. 10).
Лимфангиоэктазия кишечника характеризуется диффузным или локальным расширением лимфатических сосудов кишечника в слизистой, подслизистой или в субсерозном слое. Лимфа, богатая белками, липидами и лимфоцитами, просачивается в просвет кишечника, что приводит к ЭПБ, стеаторее и лимфоцитопении. Часто возникают гипоальбуминемия, гипогаммаглобулинемия, отеки, лимфоцитопения, нарушение всасывания жиров и жирорастворимых витаминов, а также хилезный асцит. Лимфангоэктазия кишечника также может проявляться асцитом, периферическими отеками и низким уровнем сывороточного альбумина. Этиология первичной лимфангиоэктазии кишечника неизвестна.
Несколько генов, включая рецептор 3 фактора роста эндотелия сосудов (VEGFR3), просперо-гомеобокс-транскрипционный фактор (PROX1), транскрипционные факторы (FOXC2) и SRY (область, определяющая пол Y) — Бокс 18 (SOX18), участвуют в развитии лимфатической системы и, как было показано, изменяют экспрессию в слизистой оболочке ДПК у пациентов с лимфангиоэктазией кишечника.
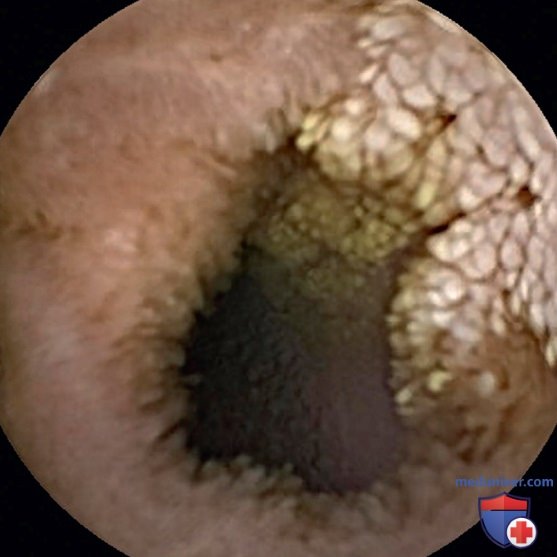
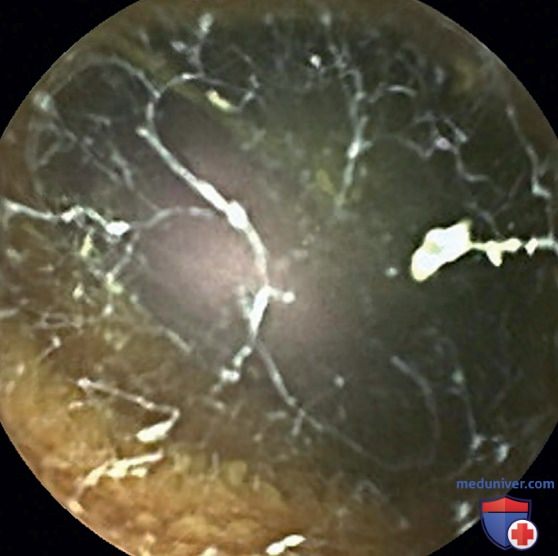
Недавно как причина первичной ЭПБ была описана мутация в CD55, в регуляторе активации комплемента. Диагноз ЭПБ подтверждается типичными клиническими и лабораторными данными в сочетании с повышенным уровнем α1-антитрипсина в кале. Рентгенологические признаки (равномерное, симметричное утолщение складок слизистой оболочки по всей тонкой кишке) характерны, но неспецифичны. Биопсия слизистой оболочки тонкой кишки у пациентов с лимфангиоэктазией кишечника может выявить расширенные лимфатические капилляры с деформацией ворсинок и отсутствием воспалительной инфильтрации. Пятнистый характер изменений и более глубокое поражение слизистой иногда приводят к л/о-результатам биопсии тонкой кишки. Видеокапсульная эндоскопия может выявить аналогичные поражения (рис. 2 и 3).
Рисуснок 2. Утолщенные ворсинки, обнаруженные при видеокапсульной эндоскопии в проксимальном отделе подвздошной кишки.
Рисуснок 3. Богатые белком агрегаты лимфатической жидкости, обнаруженные с помощью видеокапсульной эндоскопии в просвете кишечника.
Лечение ЭПБ, как правило, является поддерживающим и состоит из диеты с низким содержанием жира и высоким содержанием белка. Пациентам с лимфангиоэктазией кишечника рекомендуется диета с низким содержанием жира и высоким содержанием белка, дополненная среднецепочечными триглицеридами. Для того чтобы избежать осложнений отека, помимо корректировки рациона питания, необходимо лечение, направленное на основную причину заболевания, а также поддерживающая терапия. Парентеральное питание требуется редко. Если поражентолько участок кишечника, может быть рассмотрено выполнение хирургической резекции. У нескольких пациентов с лимфатическими мальформациями и генерализованными аномалиями лимфатической системы с успехом применяли пропранолол. Сообщалось об успешном применении ингибитора mTOR эверолимуса у пациента с первичной лимфангэктазией кишечника. Прогноз зависит от тяжести и вариантов лечения основного заболевания.