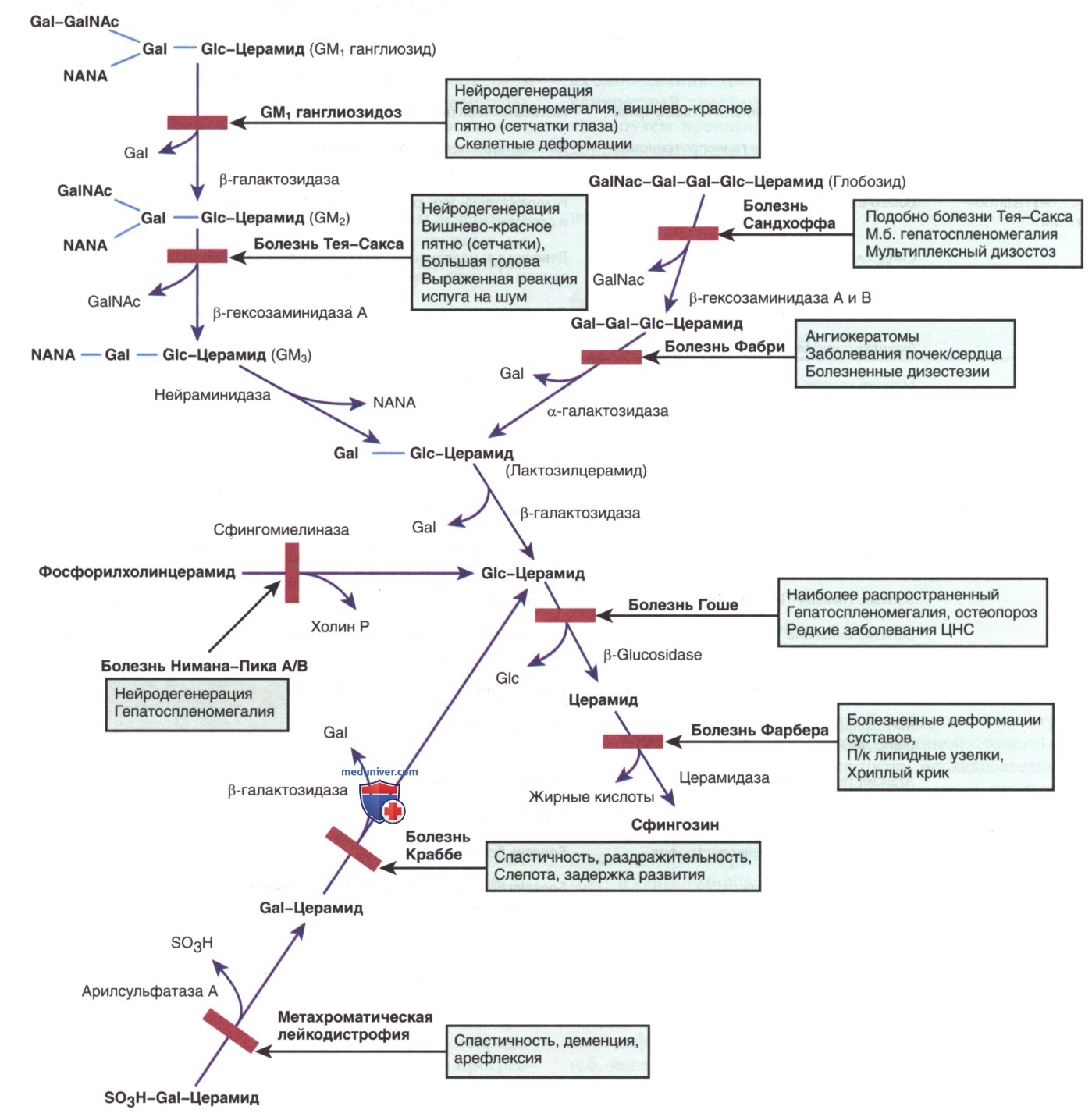
Сфинголипидозы характеризуются в/клеточным накоплением липидных субстратов в результате дефектного катаболизма сфинголипидов, входящих в состав клеточных мембран (рис. 1). Сфинголипидозы подразделяются на шесть видов: болезнь Нимана-Пика, болезнь Гоше, ганглиозидоз GM1, ганглиозидоз GM2, болезнь Краббе и метахроматическая лейкодистрофия. Болезнь Нимана-Пика и болезнь Гоше обсуждаются в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
Рисунок 1. Путь деградации сфинголипидов, показывающий участки дефицита ферментов и связанные с ними нарушения. Сфинголипиды состоят из церамидной основы с олигосахаридными боковыми цепями. Gal- - галактозо-; GalNAc — N-ацетил-галактозы; Glc- - глюкозил-; NANA — N-ацетилнейраминовая кислота
а) Ганглиозидозы. Ганглиозиды — гликосфинголипиды, нормальные составляющие нейрональных и синаптических мембран. Основная структура ганглиозида GM1 состоит из олигосахаридной цепи, присоединенной к гидроксильной группе церамида и сиаловой кислоты, связанной с галактозой. Ганглиозиды катаболизируются путем последовательного расщепления молекул сахара специфическими экзогликозидазами. Нарушения в катаболизме приводят к накоплению ганглиозида внутри клетки. Дефекты деградации ганглиозидов можно разделить на две группы: ганглиозидозы GM1 и ганглиозидозы GM2.
1. GM1 ганглиозидозы. Классификация ганглиозидозов GM1 на три подтипа основана на возрасте дебюта: инфантильный (тип 1), ювенильный (тип 2) и взрослый (тип 3). Это состояние имеет АуР-тип наследования и является результатом выраженного дефицита кислотной β-галактозидазы. Этот фермент м.б. исследован в лейкоцитах и культивируемых фибробластах. Ген кислотной β-галактозидазы был идентифицирован на хромосоме 3p22.3. Пренатальная диагностика возможна путем измерения кислотной β-галактозидазы в культивируемых амниотических клетках/прямого молекулярного тестирования.
Инфантильный ганглиозидоз GM1 проявляется при рождении/в неонатальном периоде анорексией, плохим сосанием и недостаточным набором МТ. Отмечается задержка развития и генерализованные судорожные приступы. Фенотип яркий и имеет много общих черт с синдромом Херлера (Hurler). Черты лица грубые, лоб выпуклый, переносица вдавлена, язык большой (макроглоссия), десны гипертрофированы. Гепатоспленомегалия наблюдается в начале заболевания в результате накопления пенистых гистиоцитов, а кифосколиоз проявляется вследствие формирования переднего клюва тел позвонков.
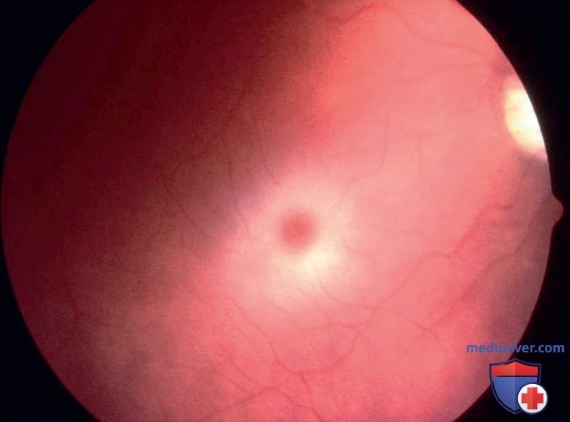
При неврологическом обследовании преобладают апатия, прогрессирующая слепота, глухота, спастическая тетраплегия и децеребрационная ригидность. Вишнево-красное пятно в макулярной области визуализируется в ~50% случаев. Оно характеризуется наличием непрозрачного кольца (сфинголипидные ганглиозные клетки сетчатки), окружающего нормальную красную ямку (рис. 2). Дети редко доживают до 2-3 лет, смерть может наступить от аспирационной пневмонии.
Рисунок 2. Вишнево-красное пятно у пациента с ганглиозидозом GM1. Обратите внимание на белое кольцо сфинголипидных ганглиозных клеток, окружающих ямку.
Ювенильный ганглиозидоз GM1 имеет отсроченное начало с дебютом в ~1 год. Начальные симптомы состоят из нарушения координации, слабости, атаксии и регрессии речи. Позже появляются судороги, спастичность, децеребрационная ригидность и слепота. В отличие от инфантильного, для этого типа обычно не характерны грубые черты лица и гепатоспленомегалия. Рентгенографическое исследование поясничных позвонков может выявить незначительные изменения. Дети редко доживают до 10 лет.
2. GM2 ганглиозидозы. Ганглиозидозы GM2 представляют собой гетерогенную группу АуР-наследуемых расстройств, представленных несколькими подтипами, включая болезнь Тея-Сакса, болезнь Сандхоффа, ювенильный ганглиозидоз GM2 и взрослый ганглиозидоз GM2. Болезнь Тея-Сакса наиболее распространена среди еврейского населения ашкенази и в США имеет частоту носителей 1:30 евреев. Болезнь Тея-Сакса вызывается мутациями в гене НЕХА, расположенном на хромосоме 15q23.
Младенцы кажутся здоровыми до 6 мес, за исключением выраженной реакции испуга на шум, которая проявляется вскоре после рождения. Затем заболевшие дети начинают отставать в развитии и к 1 году теряют способность стоять, сидеть и говорить. Ранняя гипотония перерастает в прогрессирующую спастичность, за которой следует неуклонное ухудшение состояния, сопровождающееся судорогами, слепотой, глухотой и вишнево-красными пятнами на глазном дне почти у всех пациентов (см, рис. 2).
Макроцефалия становится очевидной к 1 году и является результатом 200-300-кратного увеличения содержания ганглиозида GM2, депонированного в ГМ. Немногие дети живут дольше 3-4 лет, и смерть обычно вызвана аспирацией/брон-хопневмонией. В тканях больных болезнью Тея-Сакса обнаружен дефицит изофермента гексозаминидазы А. Существует точный и недорогой тест на обнаружение носителей (сывороточная/лейкоцитарная гексозаминидаза А), который является эффективным инструментом в определенной группе евреев-ашкенази. Целенаправленный скрининг способствовал тому, что в настоящее время в США редко рождаются дети с данной патологией и чаще — у родителей не евреев, не проходивших скрининг.
Болезнь Сандхоффа похожа на болезнь Тея-Сакса по способу проявления, включая прогрессирующую потерю двигательных и языковых навыков, начинающуюся в 6 мес. Судороги, вишнево-красные пятна на глазном дне, макроцефалия и «кукольное лицо» отмечены у большинства пациентов; однако у детей с болезнью Сандхоффа м.б. спленомегалия. Зрительные вызванные потенциалы в пределах нормы в начале обоих заболеваний, но становятся аномальными/отсутствуют по мере прогрессирования. Слуховые реакции ствола ГМ обнаруживают длительные задержки. Диагноз болезни Сандхоффа устанавливается путем обнаружения снижения уровня гексозаминидаз А и лейкоцитов в сыворотке крови. Дети обычно умирают к 3 годам. Болезнь Сандхоффа вызвана мутациями в гене НЕХВ, расположенном на хромосоме 5q13.
Ювенильный ганглиозидоз GM2 дебютирует в среднем детском возрасте, первоначально проявляется неуклюжестью, затем атаксией. Постепенно развиваются признаки спастичности, атетоза, нарушения речи и судорог. Прогрессирующая потеря зрения вызвана атрофией зрительного нерва, но вишнево-красные пятна на глазном дне редко встречаются при данной форме ганглиозидоза GM2. Дефицит гексозаминидазы у этих пациентов вариабелен (общий дефицит близок к норме). Смерть наступает в ~15 лет.
Взрослый ганглиозидоз GM2 характеризуется множеством неврологических признаков, включая медленно прогрессирующую атаксию, спастичность, дистонию, атрофию проксимальных мышц и дизартрию. Как правило, острота зрения и интеллектуальные функции не нарушаются. Активность только гексозаминидазы А/гексозаминидаз А и В и лейкоцитов в сыворотке крови значительно снижены.
б) Болезнь Краббе (глобоидно-клеточная лейкодистрофия). Болезнь Краббе — редкое АуР-нейродегенеративное заболевание, характеризующееся значительной потерей миелина и наличием глобоидных тел в белом в-ве. Ген болезни Краббе (GALC) расположен на хромосоме 14q24.3-q32.1. Заболевание возникает в результате выраженного дефицита лизосомального фермента галактоцереброзида β-галактозидазы (GALC). Болезнь Краббе — это расстройство, связанное с разрушением миелина, а не его аномальное образование. В норме миелинизация начинается в III триместре, что соответствует быстрому повышению активности GALC в ГМ.
У пациентов с болезнью Краббе галактоцереброзид не может метаболизироваться во время нормального обмена миелина из-за дефицита GALC. При введении галактоцереброзида в ГМ экспериментальных животных происходит реакция глобоидных клеток. Считается, что подобное явление наблюдается и у человека; неметаболизированный галактоцереброзид стимулирует образование глобоидных клеток, отражающих разрушение олигодендроглиальных клеток. Поскольку олигодендроглиальные клетки ответственны за выработку миелина, их потеря приводит к разрушению миелина. Т.о., производится дополнительный галактоцереброзид и формируется порочный круг разрушения миелина.
Симптомы заболевания становятся очевидными в первые несколько месяцев жизни и включают чрезмерную раздражительность и плач, необъяснимые эпизоды гиперпирексии, рвоту и трудности с питанием. В начальной стадии болезни Краббе детей часто лечат от колик/аллергии на молоко с частыми изменениями тактики. Генерализованные судороги могут появиться на ранних стадиях заболевания. Изменения в тонусе с ригидностью и опистотонусом, зрительной невнимательностью в результате оптической атрофии становятся очевидными по мере прогрессирования заболевания.
На более поздних стадиях заболевания основными симптомами являются слепота, глухота, отсутствие глубоких сухожильных рефлексов и децеребрационная ригидность. Большинство пациентов умирают <2 лет. МРТ и MP-спектроскопия информативны при оценке степени демиелинизации. Трансплантация пуповинной крови (стволовых клеток) от неродственных доноров у бессимптомных детей может изменить прогноз на более благоприятный, но не поможет пациентам с неврологическими симптомами.
Поздняя стадия болезни Краббе дебютирует в детском/подростковом возрасте. У пациентов наблюдается атрофия зрительного нерва и кортикальная слепота, и их состояние можно спутать с адренолейкодистрофией. Заметны медленно прогрессирующие нарушения походки, в т.ч. спастичность и атаксия. Как и в случае классического заболевания, глобоидные клетки в изобилии присутствуют в белом в-ве, а в лейкоцитах отмечается дефицит GALC. Исследование СМЖ указывает на повышенное содержание белка, а скорость нервной проводимости заметно замедляется в результате сегментарной демиелинизации периферических нервов.
в) Метахроматическая лейкодистрофия. Это нарушение метаболизма миелина имеет АуР-тип наследования и характеризуется дефицитом активности арилсульфатазы А. Ген ARSA расположен на хромосоме 22q13.33. Отсутствие/дефицит арилсульфатазы А приводит к накоплению цереброзидсульфата в миелине как в ЦНС, так и в периферической НС вследствие неспособности расщеплять сульфат из галактозил-3-сульфатцерамида. Считается, что избыток цереброзида сульфата вызывает распад миелина. Пренатальная диагностика метахроматической лейкодистрофии проводится путем определения активности арилсульфатазы в ворсинках хориона/культивируемых клетках околоплодных вод.
Крезиловый фиолетовый, нанесенный на образцы тканей, вызывает метахроматическое окрашивание сульфатидных гранул, что дает болезни ее название. Некоторые люди с низкой активностью фермента арилсульфатазы А клинически здоровы и имеют псевдодефицитное состояние, которое м.б. подтверждено только дополнительными генетическими/биохимическими тестами. Формы метахроматической лейкодистрофии обычно классифицируются в зависимости от возраста начала заболевания: поздний инфантильный, ювенильный и взрослый.
Поздняя инфантильная метахроматическая лейкодистрофия начинается с нарушений походки в 1-2 года. Сначала ребенок выглядит неуклюжим и часто падает, но постепенно его подвижность значительно снижается, и для ходьбы требуется поддержка. Конечности гипотоничны, а глубокие сухожильные рефлексы отсутствуют/ослаблены. В течение следующих нескольких месяцев ребенок уже не может стоять, и снижение интеллектуальных функций становится очевидным. Речь невнятная и дизартричная, ребенок выглядит умственно отсталым и апатичным. Зрительная фиксация ослаблена, имеется нистагм, а исследование сетчатки показывает атрофию зрительного нерва. В течение 1 года от начала заболевания ребенок не может сидеть без поддержки, присоединяются прогрессирующие декортикальные позы.
Кормление и глотание нарушаются из-за псевдобульбарных параличей, требуется постановка гастростомы. Состояние пациентов резко ухудшается, и они умирают от аспирации/бронхопневмонии в 5-6 лет. Нейрофизиол. диагностика демонстрирует замедление скорости периферической нервной проводимости и прогрессирующие изменения при тестировании зрительных вызванных потенциалов, слуховых реакциях ствола ГМ и соматосенсорных вызванных потенциалов. КТ и МРТ ГМ указывают на диффузную симметричную атрофию мозжечка и белого в-ва, в СМЖ повышен белок. Трансплантация костного мозга/генная терапия лентивирусными гемопоэтическими стволовыми клетками является перспективной экспериментальной терапией для лечения поздних инфантильных форм при выявлении их в дебюте заболевания.
Ювенильная метахроматическая лейкодистрофия имеет много общих черт с предыдущей формой, но начало симптомов задерживается до 5-10 лет. Снижение успеваемости в школе и личностные изменения являются предвестниками заболевания. Отмечается нарушение координации походки, недержание мочи и дизартрия. Повышается мышечный тонус, может наблюдаться атаксия, дистония и тремор. В терминальных стадиях появляются генерализованные тонико-клонические судороги, поддающиеся лечению. Пациенты редко доживают до середины подросткового возраста.
У взрослых метахроматическая лейкодистрофия встречается со второй по шестую декады жизни. Нарушения памяти, психические расстройства и личностные изменения являются характерными чертами заболевания. Медленно прогрессирующие неврологические симптомы, включая спастичность, дистонию, атрофию зрительного нерва и генерализованные судороги, приводят к прикованному к постели состоянию, характеризующемуся декортикацией и отсутствием контакта с окружающими.