Аутоиммунный энцефалит включает обширную группу клинических синдромов, которые могут возникать в любом возрасте (от 1 года до зрелого возраста), но преимущественно у молодых людей и детей (табл. 8). Некоторые из этих нарушений обусловлены образованием АТл к поверхностным белкам нейрональных клеток и синаптических рецепторов, участвующих в синаптической передаче, пластичности и возбудимости нейронов.
Синдромы варьируют в зависимости от вида АТл, а фенотипы напоминают те, в которых функция АГн была фармакологически/генетически модифицирована.
Большинство из этих расстройств являются тяжелыми и потенциально смертельными, но пациенты часто хорошо отвечают на иммунотерапию. Кроме того, из-за широкого спектра симптомов, включая изменения поведения, психоз, кататонию, бессонницу, дефицит памяти, судороги, аномальные движения и вегетативную дисрегуляцию, пациенты обычно нуждаются в мультидисциплинарном подходе к лечению, часто в ОРИТ.
Идентификация этих нарушений обеспечивает постановку окончательного диагноза для многих случаев энцефалита, ранее считавшихся идиопатическими, инфекционными/постинфекционными, даже если возбудители не были обнаружены. Поскольку этиология и патогенетические механизмы были неизвестны, некоторые из этих расстройств ранее определялись описательными терминами.
Более половины случаев с неопределенным названием «летаргический энцефалит» и некоторые случаи хореоатетоза при постгерпетическом простом энцефалите, как теперь известно, являются энцефалитом с анти-N-метил-D-аспартатным рецептором (NMDAR).
Механизмы, запускающие выработку АТл, неизвестны. В небольшой подгруппе подростков/молодых взрослых пациентов наличие опухоли, экспрессирующей целевой нейрональный АГн, вероятно, способствует запуску иммунного ответа. Кроме того, высокая распространенность продромальных вирусоподобных симптомов позволяет предположить, что неспецифические вирусные инфекции могут способствовать нарушению иммунной толерантности к нейрональным белкам и повышению проницаемости ГЭБ для АТл.
Тем не менее при многих из этих заболеваний ГЭБ кажется неповрежденным, и есть доказательства того, что ауто-АТл синтезируются в ЦНС плазматическими клетками, которые образуют часть местных мозговых и менингеальных воспалительных инфильтратов.
а) Анти-N-метил-D-аспартат рецепторный энцефалит. При этом заболевании мишенью АТл класса IgG являются субъединицы GluN1 NMDA-рецепторов. Точная частота этого заболевания неизвестна, но оно считается второй по частоте причиной аутоиммунного энцефалита после острого диссеминированного энцефаломиелита у детей и подростков. В целом заболевание преобладает у женщин (80%), хотя у пациентов <12 лет чаще встречается у мальчиков (40%). Возникающий в результате симптомокомплекс очень предсказуем и обычно развивается поэтапно.
У подростков и молодых людей это расстройство обычно проявляется ярко выраженными психическими отклонениями, которые могут включать быстро прогрессирующую тревогу, возбуждение, бредовые мысли, странное поведение, лабильный аффект, расстройства настроения (мания), кататонические черты, дефицит памяти, дезинтеграцию языка, агрессию и бессонницу/др. нарушения сна. Во многих случаях этим симптомам предшествует несколько дней продромальной головной боли, лихорадки и симптомов вирусной инфекции. Пациентам часто ошибочно диагностируется первичный психоз/первичное психическое расстройство.
Однако через несколько дней/недель появляются дополнительные симптомы, включая снижение уровня сознания, судороги (возможен эпилептический статус), дискинезии конечностей/оральные дискинезии, хореоатетоидные движения и вегетативную нестабильность (тахикардию, брадикардию, колебания АД, гиповентиляцию, гипертермию и сиалорею). В редких случаях возникают брадикардия и сердечные паузы, иногда требующие кратковременного использования кардиостимулятора.
Это расстройство может встречаться у детей раннего возраста и младенцев (самому младшему пациенту, идентифицированному на сегодняшний день, было 2 мес), и, хотя динамика синдрома аналогична эволюции взрослых, у молодых пациентов чаще наблюдаются судороги и двигательные расстройства. Из-за возраста пациентов психиатрически-поведенческие признаки м.б. пропущены. В младшей возрастной группе изменения в поведении включают раздражительность, приступы гнева, возбуждение, агрессию, снижение речевой функции, мутизм и аутистическую регрессию. Более того, в отличие от взрослых, у некоторых детей развиваются мозжечковая атаксия и гемипарез; напротив, вегетативная дисфункция у детей обычно протекает более мягко и менее тяжело.
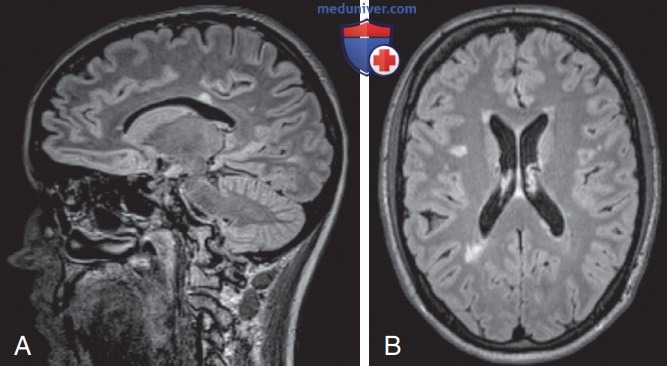
При проведении МРТ ГМ выявляется патология у 35% пациентов, обычно это неспецифические кортикальные и подкорковые нарушения на T2-FLAIR, иногда с преходящим кортикальным/менингеальным усилением; могут возникать неспецифические нарушения со стороны белого в-ва. Однако если преобладают изменения белого в-ва, то следует заподозрить смежный синдром (рис. 1 и 2).
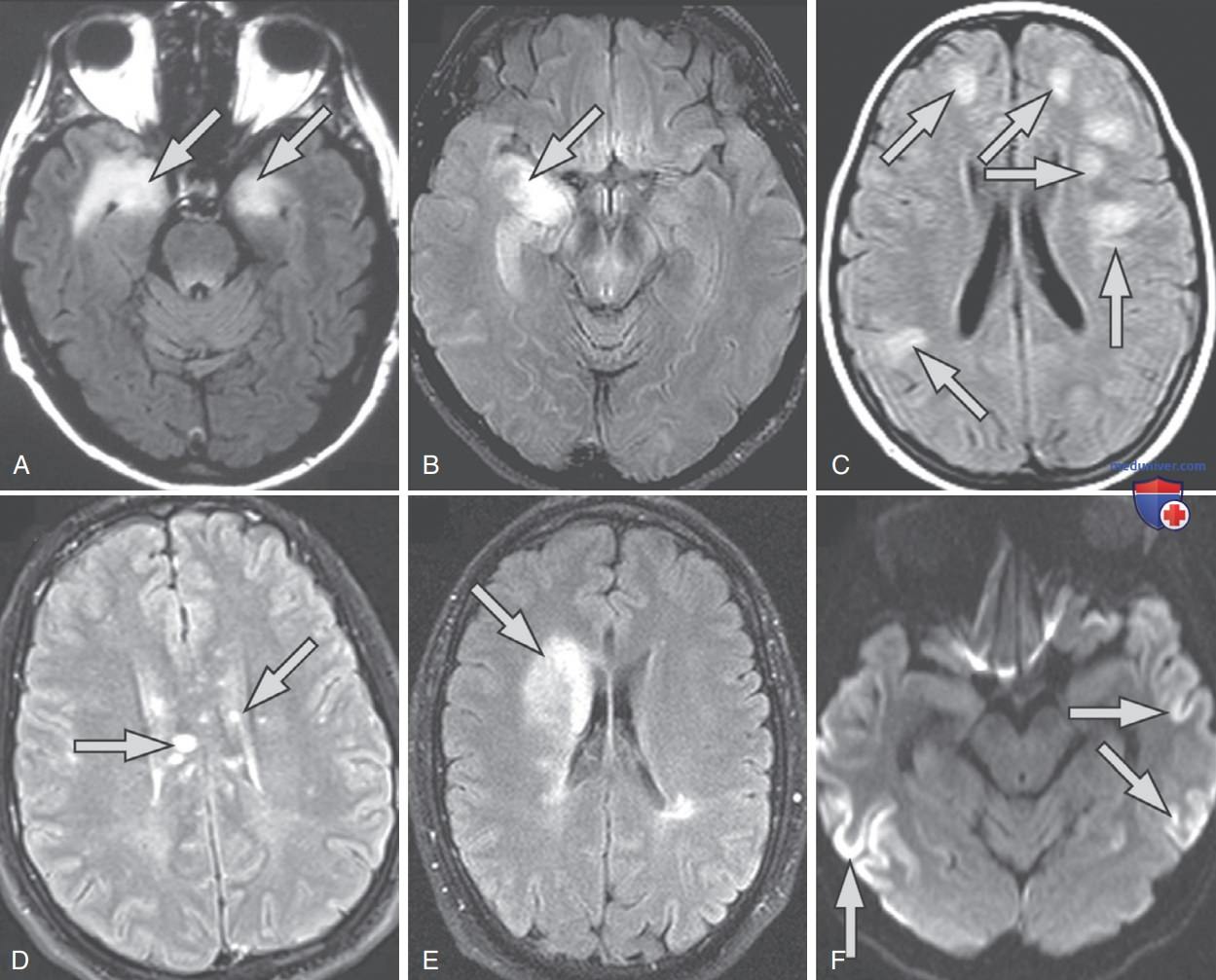
Рисунок 1. Схожие демиелинизирующие синдромы у двух пациенток с энцефалитом анти-N-метил-D-аспартатного рецептора: A — гиперинтенсивные поражения мозолистого тела в режиме восстановления инверсии с ослаблением жидкости у 20-летней пациентки с энцефалитом анти-N-метил-D-аспартатного рецептора, одно поражение в виде «пальца Доусона (Dawson)», типичное для рассеянного склероза. В последующем демиелинизация наблюдалась в перивентрикулярном белом в-ве, причем в двух из них отмечалось контрастное усиление. Очаги поражения были обнаружены во время последующей рутинной магнитно-резонансной томографии; однако пациентка жаловалась на усталость в течение последних 6 мес перед магнитно-резонансной томографией. Вскоре после исследования у нее развилась гипестезия обеих голеней и дисфункция мочевого пузыря. Начато лечение в/в глюкокортикостероидами, симптомы частично исчезли; B — гиперинтенсивные очаги поражения в режиме восстановления инверсии с ослаблением жидкости в перивентрикулярном белом в-ве у 26-летней пациентки с энцефалитом анти-N-метил-D-аспартатного рецептора. В общей сложности обнаружено 14 супратенториальных поражений, два из которых продемонстрировали контрастное усиление. Магнитно-резонансная томография проведена в связи с жалобой пациентки на периодическое двоение в глазах. Пациентку лечили в/в глюкокортикостероидами, двоение в глазах купировано.
Рисунок 2. Магнитно-резонансные паттерны при аутоиммунном энцефалите и его имитаторах. Типичная магнитно-резонансная томография лимбического энцефалита (A) с двусторонними аномалиями в медиальной височной доле на Т2-восстановленной инверсии с ослаблением жидкости; у этого пациента с доказанным аутопсией лимбическим энцефалитом не было сывороточных/-антинейронных антител центральной нервной системы. Пациент с окончательным диагнозом глиомы (B), у которого наблюдалось одностороннее поражение правого гиппокампа, имитирующее лимбический энцефалит. Типичная магнитно-резонансная томография острого диссеминированного энцефаломиелита (C) с двусторонними крупными очагами поражения белого в-ва. Множественные поражения мозолистого тела у пациента с синдромом Сусака (Susac) (D). Магнитно-резонансная томография пациента с перекрестным синдромом (антитела к NMDA-рецептору и миелиновому олигодендроцитарному гликопротеину) (E) демонстрирует правую лобную аномалию, характерную для демиелинизации. Диффузионная последовательность магнитно-резонансной томографии у пациента с α-амино-3-гидрокси-5-метилизоксазол-4-пропионат-рецепторным антител-ассоциированным энцефалитом (F) имитирует изменения магнитно-резонансной томографии, наблюдаемые у пациентов с болезнью Крейтцфельдта-Якоба (Creutzfeldt, Jakob). Левая сторона изображений = правая сторона головного мозга.
СМЖ изначально аномальна у 80% пациентов, при этом выявляется умеренный лимфоцитарный плеоцитоз и, реже, повышение уровня белка и олигоклональные полосы. ЭЭГ патологическая практически у всех пациентов, она обычно демонстрирует очаговую/диффузную медленную активность в δ-/θ-диапазонах, которая с движениями не связана. Кроме того, у многих пациентов развивается эпилептическая активность, требующая проведения видеомониторинга для последующего адекватного клинического ведения.
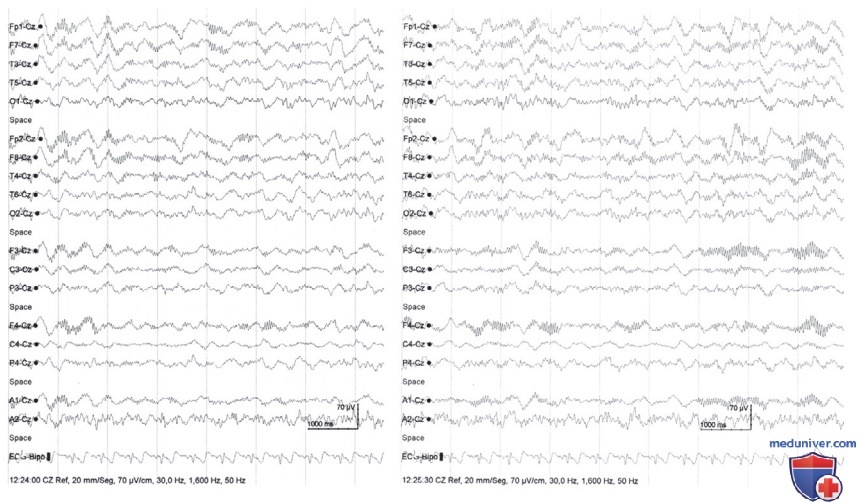
Характерный паттерн ЭЭГ, называемый экстремальной δ-щеткой, характеризующийся β-δ-комплексами, встречается у 30% взрослых и был описан у детей (рис. 3).
Рисунок 3. Электроэнцефалография, демонстрирующая паттерн, называемый экстремальной δ-щеткой, у 14-летней девочки с энцефалитом анти-N-метил-D-аспартатного рецептора. Обнаружено, что этот паттерн характерен для антиэнцефалита анти-N-метил-D-аспартатного рецептора. Он состоит из почти непрерывной комбинации δ-активности с наложением быстрой активности, обычно в β-диапазоне, симметрично охватывающей все области, с преобладанием во фронтальной, у пациентов при отсутствии седации/анестезии.
Диагноз устанавливается путем выявления АТл NMDAR в ликворе и сыворотке крови. Вероятность их обнаружения выше для СМЖ по сравнению с сывороткой крови (100% против 85%), и уровни АТл в ликворе, по-видимому, лучше коррелируют с исходом. АТл у пациентов могут оставаться, хотя и в более низких титрах, после выздоровления.
Наличие лежащей в основе опухоли, обычно тератомы, зависит от возраста и пола. В то время как 40% девочек >12 лет имеют основную тератому яичника, наличие опухоли является исключительным у мальчиков и девочек/молодых взрослых пациентов мужского пола. У детей МРТ ОБП и ОМТ, а также УЗИ БП и яичек являются предпочтительными методами скрининга опухоли.
У небольшого числа пациентов анти-NMDAR-энцефалит возникает одновременно с/после инфицирования разл. патогенами, включая Mycoplasma pneumoniae, ВПГ 1-го типа, ВГЧ 6, энтеровирус и вирус гриппа. За исключением ВПГ 1-го типа, патогенная связь с большинством этих инфекций не установлена. Имеются данные, что у некоторых пациентов с ВПГ-энцефалитом развиваются АТл против субъединицы GluN1 NMDAR и др. белков и рецепторов поверхности нейрональных клеток, что приводит к появлению новых/рецидивирующих неврологических симптомов через 2-12 нед после завершения лечения ВПГ-энцефалита. У детей <4 лет этот тип аутоиммунного энцефалита обычно проявляется хореоатетозом и дискинезиями (известными как хореоатетоз после ВПГ-энцефалита).
Напротив, у детей старшего возраста и взрослых чаще развиваются преимущественно поведенческие аномалии.
Хотя никаких проспективных клинических исследований не проводилось, есть доказательства, что удаление опухоли в случае необходимости и своевременная иммунотерапия улучшают результат. Большинство детей получают иммунотерапию первой линии, включая ГКС, в/в Ig, плазмаферез. Однако, поскольку эти методы лечения терпят неудачу почти у 50% пациентов, а также с увеличением числа сообщений об эффективности ритуксимаба, он все чаще применяется в комбинации с в/в Ig и ГКС/после иммунотерапии первой линии. Циклофосфамид м.б. эффективен при отсутствии ответа на выше указанные методы лечения.
Хотя смертность от анти-NMDAR-энцeфaлитa составляет 7%, 80% пациентов имеют существенное улучшение/выздоравливают полностью. Выздоровление обычно происходит медленно и может занять до 2 лет после появления симптомов. Последними симптомами, требующими лечения, являются проблемы в социальных взаимодействиях, общении и исполнительных функциях. Рецидивы встречаются у 15% пациентов; они могут развиваться как парциальные синдромы, обычно протекают мягче, чем первый эпизод, и одинаково хорошо поддаются иммунотерапии. Ранняя комплексная иммунотерапия, по-видимому, предотвращает/уменыпает количество рецидивов. Эффективность постоянной иммуносупрессии такими ЛП, как азатиоприн/микофенолата мофетил, в профилактике рецидивов неизвестна.
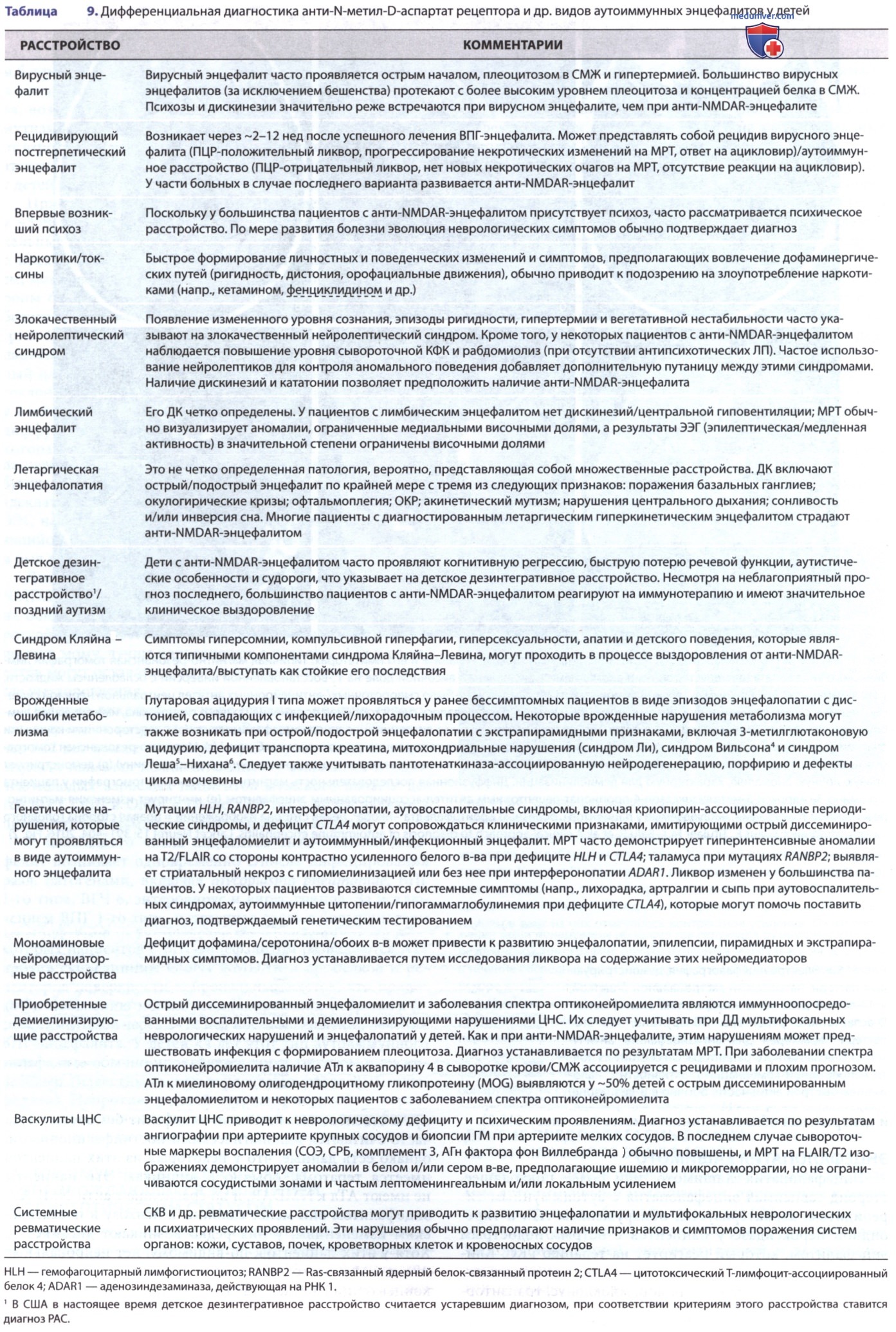
ДД анти-NMDAR-энцефалита обширна и варьирует в зависимости от стадии заболевания (табл. 9). Наиболее часто рассматриваемыми расстройствами являются вирусный энцефалит, нейролептический злокачественный синдром, острый психоз и злоупотребление наркотиками.
б) Другие виды энцефалита с участием аутоантител, направленных против нейрональных антигенов клеточной поверхности. Энцефалит с АТл против рецептора ГАМК — это редкий аутоиммунный энцефалит, который может поражать детей (40% пациентов <18 лет) и сопровождается эпилептическим статусом, рефрактерными припадками, эпилепсией partialis continua в ассоциации с АТл против α1, β3 и γ2 субъединиц ГАМК. У маленьких детей могут развиться патологические движения, имитирующие анти-NMDAR-энцефалит, но при «-» результатах на АТл к NMDAR.
В отличие от др. типов аутоиммунного энцефалита, при которых МРТ ГМ обычно в норме/выявляет неспецифические изменения, у взрослых и детей с этим расстройством часто развиваются мультифокальные гиперинтенсивные корково-подкорковые нарушения на FLAIR/T2. У взрослых этот энцефалит может протекать на фоне тимомы, но у детей редко бывает данная опухоль.
Синдром Офелии — это форма энцефалита, которая возникает в сочетании с лимфомой Ходжкена и преимущественно поражает молодых взрослых, подростков и детей. У некоторых пациентов вырабатываются АТл против mGluR5 — рецептора, участвующего в обучении и запоминании. Неврологические симптомы очень чувствительны к лечению опухоли и иммунотерапии.
Аутоиммунный лимбический энцефалит представляет собой воспалительный процесс в лимбической системе, включая медиальные височные доли, миндалину и поясную извилину. У взрослых наиболее часто иммунноопосредованный лимбический энцефалит возникает в сочетании с АТл против, как ранее считались, напряженно-зависимых калиевых каналов (VGKCs), но на самом деле нацеленных на секретируемый нейрональный белок, называемый богатым лейцином, глиома-инактивированным 1 белком (LGI1), и белок Caspr2, экспрессирующийся в ГМ и юкстапаранодальных областях миелинизированных нервов.
У пациентов с лимбическим энцефалитом с АТл к LGI1 часто развивается гипонатриемия; у некоторых пациентов этому расстройству предшествуют дистонические/миоклонические движения, описываемые как фациобрахиальные дистонические приступы. У пациентов с АТл к Caspr2 может развиться лимбический энцефалит, нейромиотония и синдром Морвана (Morvan), который включает энцефалопатию, судороги, нарушение сна, вегетативную дисфункцию и нейромиотонию. Исследования показали, что у пациентов без АТл к LGI1/Caspr2 обнаружение АТл к комплексу потенциал-зависимого калиевого канала (VGKC; англ. Voltage-Gated Potassium Channels) имеет очень ограниченное клиническое значение.
У детей выявление АТл к LGI1/Caspr2 необычно; поэтому «+» тест на АТл к VGKC-комплексу следует интерпретировать с осторожностью, поскольку он не обязательно указывает на аутоиммунный энцефалит. У детей аутоиммунный/паранеопластический лимбический энцефалит встречается редко. К сожалению, любой тип энцефалопатии, приводящий к припадкам и изменению памяти и поведения, часто диагностируют как лимбический энцефалит, что делает поиск литературы с использованием термина «лимбический энцефалит» ненадежным.
За исключением пациентов с NMDAR/ГАМКAR АТл-ассоциированным энцефалитом в английской литературе зарегистрировано <30 детей с лимбическим и др. типами АТл-ассоциированным энцефалитом, некоторые из которых обусловлены АТл против рецепторов поверхности нейронов/белков (ГАМКBR, DPPX, GlyR), в/клеточных белков (Hu, Ма2, GAD65, амфифизин) и неидентифицированных в/клеточных белков (VGKC-комплексные белки). У некоторых пациентов выявлена основная опухоль, включая лейкемию, ганглионевробластому, нейробластому и мелкоклеточный рак яичника.
На практике определение типа ауто-АТл и локализации АГн-мишеней имеет значение, поскольку энцефалит, при котором АГн находятся на поверхности клетки (напр., NMDAR/ГАМКAR), лучше поддается иммунотерапии, чем тот, при котором АГн находятся внутри клетки (напр., GAD65).
в) Приобретенные демиелинизирующие синдромы с энцефалопатией. Острый диссеминированный энцефаломиелит является наиболее частым аутоиммунным энцефалитом у детей. Симптомы могут включать судороги, двигательный дефицит, атаксию и зрительную дисфункцию. АТл против MOG встречаются у 50-60% пациентов с острым диссеминированным энцефаломиелитом и имеют «-» прогностическое значение для развития рассеянного склероза у детей с первым демиелинизирующим эпизодом.
АТл к MOG также были описаны у пациентов с аутоиммунным энцефалитом и результатами МРТ, демонстрирующими преобладающее вовлечение серого в-ва (коры ГМ и глубоких структур серого в-ва).
Заболевания спектра оптиконейромиелита может проявляться в виде энцефалопатии с преимущественным поражением диэнцефальных и зон постремы. Эти пациенты часто имеют АТл к аквапорину 4/MOG. Определение этих АТл следует рассматривать у пациентов с энцефалопатией и результатами МРТ, демонстрирующими поражение богатых аквапорином четырех областей, таких как периакведуктальное серое в-во, гипоталамус, зрительные нервы и центральная область спинного мозга.
г) Энцефалопатия Хашимото. Энцефалопатия Хашимото, или более подходящее, стероид-зависимая энцефалопатия с аутоиммунным тиреоидитом, характеризуется обнаружением АТл к тиреоидной пероксидазе у пациентов с острым/подострым энцефалитом, который реагирует на терапию ГКС. Клинические признаки неспецифичны и могут включать инсультоподобные эпизоды, тремор, миоклонус, транзиторную афазию, нарушения сна и поведения, галлюцинации, судороги и атаксию.
В ликворе обычно наблюдается повышенный уровень белка с менее частым плеоцитозом. Результат ЭЭГ почти всегда с отклонением, часто включает генерализованную медленную активность.
МРТ ГМ обычно в норме, хотя может выявлять диффузные аномалии белого в-ва и оболочек, поддающиеся терапии ГКС. Поскольку АТл к тиреоидной пероксидазе встречаются у 10% бессимптомных детей (без энцефалопатии и в большинстве случаев с эутиреозом), а также м.б. обнаружены у некоторых пациентов с более релевантными АТл-ассоциированными заболеваниями, обнаружение АТл к тиреоидной пероксидазе следует рассматривать как маркер аутоиммунного ответа, а не как специфическое патогенное АТл. Наличие этих АТл не должно препятствовать обследованию на более релевантные АТл, такие как АТл к NMDAR.
д) Опсоклонус-миоклонус и другие виды стволово-мозжечкового энцефалита. Опсоклонус-миоклонус встречается у младенцев, подростков и взрослых, хотя, вероятно, представляет собой разл. заболевания и имеет разл. патогенетические механизмы. У младенцев синдром обычно развивается в первые 2 года жизни (в среднем 20 мес), и по крайней мере у 50% пациентов имеется нейробластома. У ребенка часто отмечаются раздражительность, атаксия, неустойчивость, миоклонус, тремор и слюнотечение.
Дополнительные симптомы могут включать невозможность ходить/сидеть, проблемы с речью, гипотонию и типичные особенности опсоклонуса, характеризующиеся быстрыми, хаотичными, разнонаправленными движениями глаз без саккадических интервалов. Поскольку опсоклонус может отсутствовать при появлении симптомов, у пациентов первоначально м.б. диагностирован острый мозжечковый синдром/ла-биринтит. Как правило, аномалии в СМЖ предполагают активацию В-клеток и наличие АТл против нейрональных белков, хотя идентификация специфического ауто-АГн трудна.
Иммунотерапия, включающая ГКС и в/в Ig, часто уменьшает аномальные движения глаз, но остаточные поведенческие, языковые и когнитивные проблемы сохраняются у большинства пациентов, часто требуя специального обучения. Кроме того, часто встречается бессонница и аномальная реакция на боль. Рецидивы возникают у 50% пациентов, как правило, в результате интеркуррентной инфекции/применения ЛП. Пациенты, получавшие более агрессивную иммуносупрессию (часто включая ритуксимаб), имеют лучшие результаты по сравнению с контрольными группами пациентов, его не получавших.
Задержка в лечении, по-видимому, обуславливает плохой неврологический исход; поэтому в случаях с нейробластомой удаление опухоли не должно задерживать начало иммунотерапии.
У подростков и молодых взрослых опсоклонус-миоклонус и стволово-мозжечковый энцефалит без опсоклонуса часто считаются идиопатическими/постинфекционными; однако есть данные, что у некоторых из этих пациентов имеется тератома, обычно в яичниках. Эти пациенты не имеют АТл к NMDAR, и по сравнению с анти-NMDAR-энцефалитом они менее склонны к психозу и поведенческим изменениям, у них редко возникают дискинезии. Хотя у этих пациентов, по-видимому, нет нейрональных АТл, в СМЖ часто наблюдается плеоцитоз и повышенная концентрация белка.
Идентификация этого субфенотипа опсоклонуса-миоклонуса важна, поскольку пациенты обычно полностью выздоравливают после курса иммунотерапии (ГКС, в/в Ig и/или плазмаферез) и удаления тератомы яичников, если она присутствует. Прогноз опсоклонуса-миоклонуса у подростков и молодых взрослых кажется лучше, чем у маленьких детей (с нейробластомой или без нее)/паранеопластического опсоклонуса у пожилых пациентов, обычно развивающегося на фоне рака МЖ, яичников и легких.
е) Энцефалит Бикерстаффа. Этот термин используется для описания пациентов с быстрым прогрессированием (<4 нед) двусторонней наружной офтальмоплегии, атаксии и снижением уровня сознания. Хотя эта патология чаще описывалась у взрослых, были выявлены и дети <3 лет. Большинство пациентов лечат ГКС, в/в Ig, плазмаферезом, что часто приносит хороший результат. Сывороточные АТл к Ig Gq1b обнаруживаются у 66% пациентов. Аномалии МРТ ГМ встречаются у 30% пациентов и обычно включают повышение Т2-сигнала в стволе ГМ, таламусе и мозжечке, а иногда и в белом в-ве.
У некоторых пациентов развиваются гипорефлексия и слабость конечностей с преимущественным аксональным поражением, имитирующие симптомы синдрома Миллера-Фишера (Miller, Fisher) и аксональный подтип синдрома Гийена-Барре (Guillain, Barre).
ж) Хроническое лимфоцитарное воспаление с периваскулярным накоплением контрастного вещества в мосту головного мозга, отвечающее на терапию стероидами. Хроническое лимфоцитарное воспаление периваскулярным накомплением контрастного в-ва в мосту ГМ, поддающееся лечению ГКС, это клинически и рентгенологически отчетливый понтино-преобладающий энцефаломиелит. У пациентов обычно наблюдается эпизодическая диплопия/лицевые парестезии с последующим развитием симптомов нарушения функции ствола ГМ и иногда спинного мозга. МРТ ГМ демонстрирует симметричное криволинейное усиление гадолиния, пронизывающее мост и вариабельно распространяющееся в продолговатый мозг, ножки мозжечка, мозжечок и средний мозг, а иногда и в спинной мозг.
Клиническое и рентгенологическое улучшение видно на фоне высоких доз ГКС, при этом ухудшение состояния может возникать после снижения этих доз, требуя проведения постоянной ГКС-/др. иммуносупрессивной терапии. ДД обширна и включает инфекции, приобретенные демиелинизирующие синдромы, гранулематозные заболевания, лимфому и васкулит. Для исключения этих и др. состояний может потребоваться проведение биопсии.
з) Аутоиммунные энцефалопатии, сопровождающиеся эпилепсией и эпилептическим статусом. Энцефалит Расмуссена — воспалительная энцефалопатия, характеризующаяся прогрессирующими рефрактерными очаговыми приступами, снижением когнитивных функций и очаговым неврологическим дефицитом, возникающим при постепенной атрофии одного полушария ГМ. Это расстройство часто встречается у детей 6-8 лет, хотя могут развиться у подростков и взрослых. Этиология заболевания неизвестна, поэтому предлагается множество теорий, в т.ч. наличие нейрональных АТл и Т-клеточных опосредованных механизмов, запускаемых вирусной инфекцией.
Ни один из этих механизмов не имеет удовлетворительного объяснения одностороннего вовлечения ГМ, характерного для данного расстройства. Лечение высокими дозами ГКС, применение плазмафереза и в/в Ig может уменьшить симптомы на ранних стадиях заболевания. Ритуксимаб и в/желудочковый интерферон гамма человеческий рекомбинантный оказались эффективными в нескольких единичных случаях. В небольшой группе пациентов, получавших такролимус, наблюдалось улучшение неврологических функций и более медленное прогрессирование церебральной гемиатрофии, но контроль над приступами не был достигнут.
Открытое исследование с использованием моноклонального АТл против ФНОа (адалимумаб) продемонстрировало достижение контроля приступов и сохранение когнитивных функций у ~50% пациентов. Наиболее эффективным методом, приводящим к контролю над судорогами, является функциональная гемисферэктомия, которая заключается в хирургическом разъединении связей с пораженным полушарием.
Открытие чувствительного к лечению энцефалита, опосредованного АТл против поверхностных клеток/синаптических белков, позволило предположить, что существует аутоиммунная основа для ряда разрушительных энцефалопатий с рефрактерными припадками. Некоторые четко определенные типы аутоиммунного энцефалита, такие как анти-NMDAR-/ГАМК-энцефалит, могут сопровождаться рефрактерными припадками/эпилептическим статусом. У большинства этих пациентов развиваются др. клинические признаки, позволяющие поставить диагноз, а обследование на соответствующие АТл подтверждает правильный диагноз и способствует началу иммунотерапии.
Разрушительная эпилептическая энцефалопатия, сопровождающаяся лихорадкой, также носящая название синдрома рефрактерной эпилептической энцефалопатии, предположительно является вызванным инфекцией аутоиммунным процессом из-за его двухфазного клинического течения и случайного обнаружения нейрональных АТл у нескольких пациентов. Однако отсутствие ответа на большинство методов лечения, включая иммунотерапию, а также редкая и противоречивая связь с разл. типами АТл ставят под сомнение аутоиммунный патогенез. Некоторые исследователи предполагают генетическую ошибку в метаболизме.
АТл к белкам VGKC-комплекса, отличным от LGI1 и Caspr2, описаны у некоторых детей с энцефалитом с эпилептическим статусом или без него. Учитывая, что целевые АГн, скорее всего, внутри клетки и реакция на иммунотерапию непредсказуема, значение этих АТл неясно.
и) Другие предполагаемые типы аутоиммунного энцефалита. Васкулит ЦНС и ревматические заболевания, вызываемые аутоиммунными механизмами, приводящие к развитию энцефалита, обсуждаются в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
Быстро прогрессирующее ожирение с гипоталамической дисфункцией, гиповентиляцией и вегетативной дисрегуляцией (синдром ROHHAD), как правило, поражает детей с нормальным развитием до 2-4 лет с последующим стремительным повышением аппетита, увеличением МТ и аномальным поведением (асоциальное поведение, вспыльчивость, импульсивность, заторможенность, вспышки эйфории и смеха, нарушение концентрации внимания), синдром сопровождается вегетативной дисфункцией (нарушения зрачковых реакций, тепловой регуляции, моторики ЖКТ) и центральной гиповентиляцией.
Аутоиммунная/паранеопластическая этиология синдрома ROHHAD подтверждается частой связью с опухолями нервного гребня, выявлением у некоторых пациентов генетических факторов, предрасполагающих к развитию аутоиммунитета, а также обнаружением у некоторых пациентов интратекальных олигоклональных полос и инфильтратов лимфоцитов и гистиоцитов в гипоталамусе.
Кроме того, у нескольких пациентов были описаны «+» ответы на иммунотерапию. Возможное генетическое происхождение предполагается из-за сходства этого синдрома с врожденным синдромом центральной гиповентиляции (синдром проклятия Ундины), обусловленном мутацией РНОХ2В, который проявляется в неонатальном периоде и вызывает вегетативные проблемы (болезнь Гиршпрунга), сопровождается опухолями нервного гребня (см. главу 446.2). Однако мутаций в РНОХ2В и др. генах у пациентов с ROHHAD не обнаружено.
Термин «базальный ганглиозный энцефалит» используется для описания пациентов с преимущественным/изолированным поражением базальных ганглиев. Эти пациенты, как правило, имеют нарушения движения и нервно-психические заболевания. Хотя эти клинические проявления могут иметь множественную этиологию, включая метаболические, токсические, генетические и инфекционные процессы, у некоторых пациентов определена иммуноопосредованная этиология. Клинических исследований не проводилось, но данные о случаях заболевания и небольшие неконтролируемые серии случаев описывают потенциальную эффективность иммунотерапии. У этих пациентов нечасто выявлялись АТл против рецептора дофамина-2, также как и у пациентов с хореей Сиденхэма (Sydenham) и синдромом Туретта (Tourette).
Псевдомигрень с плеоцитозом ликвора/головная боль с неврологическим дефицитом и лимфоцитозом ликвора — это плохо определенное состояние, которое преимущественно поражает молодых взрослых мужчин с семейным анамнезом мигрени, хотя могут страдать и подростки. Этот синдром характеризуется повторными эпизодами сильной головной боли с преходящим неврологическим дефицитом, сопровождается асептическим лимфоцитозом ликвора при нормальной МРТ ГМ. У пациентов часто наблюдается высокое давление ликвора, повышенная концентрация белка в ликворе и фокальное замедление ЭЭГ, которые нормализуются после эпизодов головной боли. Из-за воспалительных особенностей ликвора и высокой распространенности продромальных вирусоподобных симптомов был предложен инфекционно-аутоиммунно-опосредованный механизм.
Др. теории включают распространенную кортикальную депрессию и тригеминально-сосудистую активацию.
Иммунноопосредованный механизм и тригеминально-сосудистая активация тоже рассматриваются как возможные механизмы офтальмоплегической мигрени, также называемой рецидивирующей черепной невралгией. Это расстройство преимущественно поражает маленьких детей и характеризуется повторяющимися приступами головной боли в дополнение к параличу ЧМН III, IV и/или VI. В отличие от псевдомигрени с плеоцитозом ликвора/головной боли с неврологическим дефицитом и лимфоцитозом ликвора, исследования СМЖ не выявляют плеоцитоза, и у ~75% пациентов на МРТ обнаруживается фокальное утолщение нервов и контрастное усиление. Данные наблюдений показывают, что лечение ГКС м.б. эффективным.
При этом синдроме, как и при псевдомигрени с плеоцитозом ликвора/головной боли с неврологическим дефицитом и лимфоцитозом ликвора, ДД включает структурные, неопластические, травматические, метаболические и инфекционные нарушения.