Митохондриальные энцефаломиопатии представляют собой гетерогенную группу клинических синдромов, вызванных генетическими повреждениями, которые нарушают выработку энергии через окислительное фосфорилирование и функцию митохондрий. Признаки и симптомы этих расстройств отражают уязвимость НС, мышц и др. органов к дефициту энергии. Симптомы мозговой и мышечной дисфункции (судороги, слабость, птоз, наружная офтальмоплегия, психомоторная регрессия, потеря слуха, двигательные расстройства и атаксия) в сочетании с лактоацидозом являются характерными признаками митохондриальных нарушений. Кардиомиопатия и СД также м.б. результатом митохондриальных нарушений.
Дети с митохондриальными нарушениями часто имеют мультифокальные симптомы, которые носят интермиттирующий/рецидивирующе-ремиттирующий характер, часто в сочетании с интеркуррентными заболеваниями. Многие из этих расстройств были описаны как клинические синдромы еще до понимания их генетической основы. У детей с MELAS отмечаются задержка развития, слабость и головные боли, а также очаговые симптомы, указывающие на инсульт. При визуализации ГМ становится ясно, что повреждение не укладывается в рамки обычных сосудистых областей.
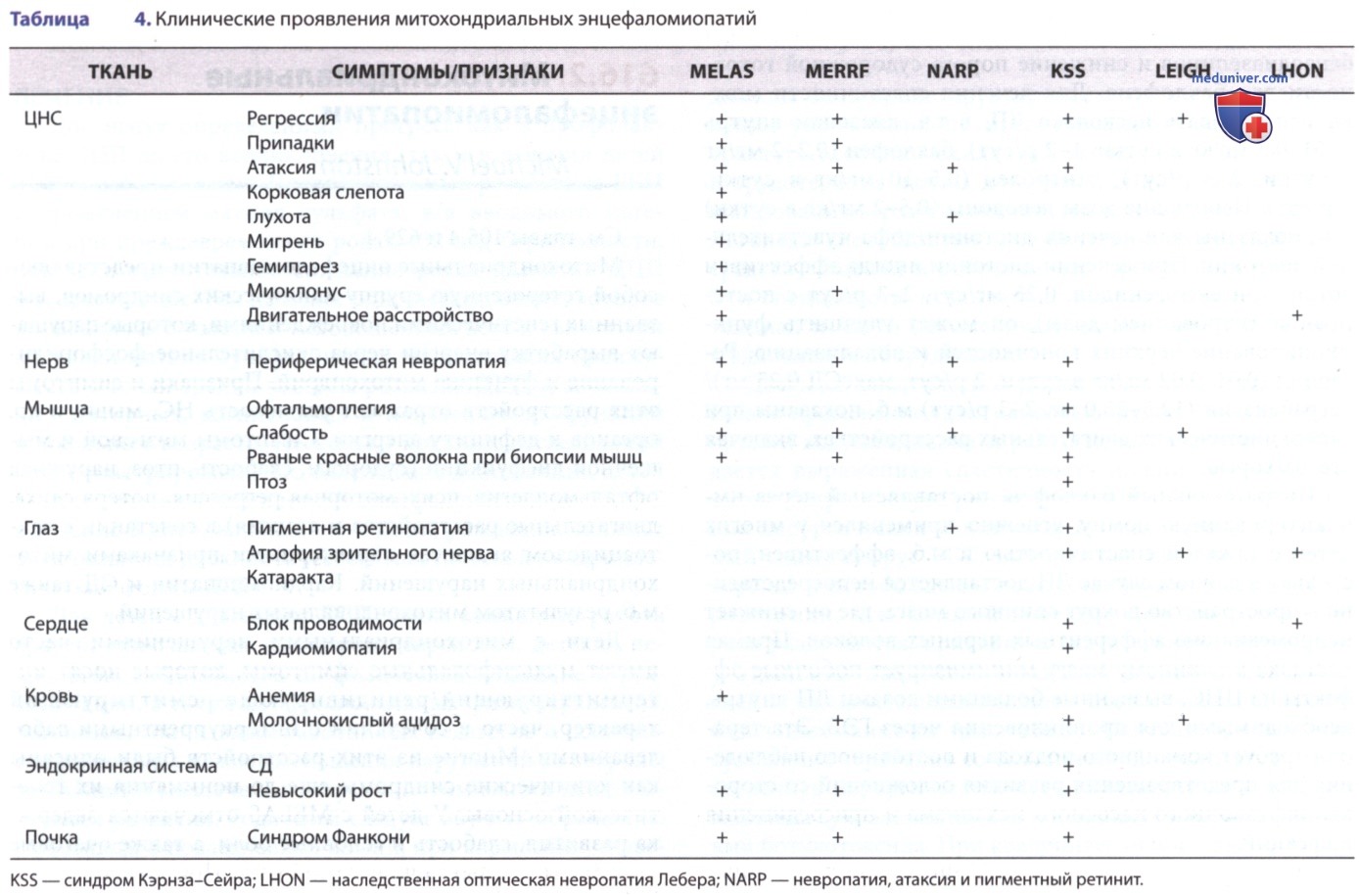
У детей с MERRF наблюдаются миоклонус и миоклонические припадки, а также периодическая мышечная слабость. Рваные красные волокна, упомянутые в названии этого расстройства, представляют собой скопления аномальных митохондрий, видимых в мышечных волокнах в срезах из биопсии мышц, окрашенных трихромным пятном Гемери (Gomori). Синдром невропатии, атаксии и пигментного ретинита, синдром Кэрнза-Сейра (Kearns, Sayre) (птоз, офтальмоплегия, блокада сердца), болезнь Ли (Leigh) (подострая некротизирующая энцефаломиелопатия) и наследственная оптическая невропатия Лебера (Leber) также являются относительно однородными клиническими подгруппами, хотя возраст выставления диагноза может варьировать (табл. 4).
Важно иметь в виду, что митохондриальные нарушения м.б. трудно диагностируемыми. Они часто представляют собой новые комбинации признаков и симптомов вследствие высокой частоты мутаций митохондриальной ДНК (мтДНК), а их тяжесть варьирует от человека к человеку. В детском возрасте чаще встречаются мутации ядерных генов (>400 возможных генов), но наблюдаются и митохондриальные генные аномалии.
Митохондриальные заболевания м.б. вызваны мутациями ядерной/мтДНК. Окислительное фосфорилирование в дыхательной цепи опосредуется четырьмя в/митохондриальными ферментными комплексами (комплексы I-IV) и двумя подвижными электронными носителями (кофермент Q и цитохром С), которые создают электрохим. протонный градиент, используемый комплексом V (АТФ-синтаза) для создания АТФ, необходимого для нормальной клеточной функции.
Поддержание окислительного фосфорилирования требует скоординированной регуляции генов ядерной и мтДНК. Человеческая мтДНК представляет собой небольшую (16,6 kb) круглую двухцепочечную молекулу, полностью секвенированную и кодирующую 37 генов, включая 13 структурных белков, все из которых являются субъединицами комплексов дыхательной цепи, а также 2 рибосомные РНК и 22 трансферные РНК, необходимые для трансляции. Ядерная ДНК отвечает за синтез ~70 субъединиц, транспортировку их в митохондрии через белки-шапероны, обеспечение их прохождения через внутреннюю мембрану митохондрий и координацию их правильной обработки и сборки. Патологию митохондриального окислительного фосфорилирования можно разделить на три группы:
1) дефекты мтДНК;
2) дефекты ядерной ДНК;
3) дефекты связи между ядерным и митохондриальным геномами.
Митохондриальная ДНК отличается от ядерной следующими пятью признаками:
1) генетическим кодом;
2) плотно упакованной информацией, т.к. не содержит интронов;
3) большей частотой спонтанных мутаций;
4) менее эффективными механизмами репарации;
5) наследованием по материнской линии.
Наследование мутаций, присутствующих в мтДНК, не относится к менделевским и м.б. довольно сложным. При оплодотворении мтДНК присутствует в сотнях/тысячах копий на клетку и передается по материнской линии от ее яйцеклетки всем ее детям, но только ее дочери могут передать ее своим детям. Благодаря процессу, называемому гетероплазмией/пороговым эффектом, мутации мтДНК м.б. неравномерно распределены между клетками в определенных тканях. Некоторые клетки получают мало/вообще не получают мутантных геномов (нормальная/дикая гомоплазмия), тогда как др. получают смешанную популяцию мутантных и диких мтДНК (гетероплазмия), а третьи получают преимущественно/исключительно мутантные геномы (мутантная гомоплазмия). Последствия материнского наследования и гетероплазмии заключаются в следующем:
1) наследование болезни происходит по материнской линии, но оба пола в равной степени подвержены этому заболеванию;
2) фенотипическая экспрессия мутации мтДНК зависит от относительных пропорций мутантных и дикого типа геномов, причем минимальное критическое число мутантных геномов необходимо для экспрессии заболевания (пороговый эффект);
3) при делении клеток пропорциональное распределение может смещаться между дочерними клетками (митотическая сегрегация), приводя к соответствующему фенотипическому изменению;
4) последующие поколения поражаются с большей частотой, чем при АуД-заболеваниях.
Критическое количество мутантных мтДНК, необходимых для порогового эффекта, может варьировать в зависимости от уязвимости ткани к нарушениям окислительного метаболизма, а также от уязвимости той же ткани с течением времени, которая может увеличиваться с возрастом. В отличие от материнских наследственных расстройств, вызванных мутациями в мтДНК, болезни, вызванные дефектами в ядерной ДНК, наследуются согласно законам Менделя. Митохондриальные заболевания, вызванные дефектами ядерной ДНК, включают дефекты транспорта субстрата (плазмалеммальный транспортер карнитина, карнитинпальмитоилтрансферазы I и II, дефекты транслоказы карнитина ацилкарнитина), дефекты окисления субстрата (комплекс пируватдегидрогеназы, пируваткарбоксилаза, дефекты в/митохондриального окисления жирных кислот), дефекты цикла Кребса (Krebs) (дефекты α-кетоглутаратдегидрогеназы, фумаразы, аконитазы) и дефекты в дыхательной цепи (комплексы I-V), в т.ч. дефекты связи окисления/фосфорилирования (синдром люфта) и дефекты транспорта митохондриальных белков.
Заболевания, вызванные дефектами мтДНК, можно разделить на вызванные точечными мутациями, унаследованными от матери (напр., синдромы наследственной оптической невропатии Лебера; MELAS; MERRF; невропатии, атаксии и пигментнго ретинита/митохондриальный синдром Ли), и вызванные делециями/дубликациями мтДНК, отражающими измененную связь между ядром и митохондриями (синдром Кэрнза-Сейра; синдром Пирсона (Pearson), редкая тяжелая энцефалопатия с анемией и дисфункцией ПЖЖ; прогрессирующая внешняя офтальмоплегия). Эти нарушения м.б. унаследованы спорадическими, АуД/АуР-механизмами, были выявлены мутации в нескольких генах, включая митохондриальную каталитическую субъединицу мтДНК-полимеразы у (POLG).
Ее мутации были выявлены у пациентов с синдромом Альперса-Гуттенлохера (Alpers, Huttenlocher), сопровождающимся рефрактерным судорожным расстройством и печеночной недостаточностью, а также при АуД/АуР прогрессирующей внешней офтальмоплегии, расстройствах спектра детской миоцереброгепатопатии, миоклонической эпилепсии, миопатии сенсорной атаксии и POLG-ассоциированные расстройства спектра нейропатии атаксии. Др. гены, регулирующие поступление нуклеотидов для синтеза мтДНК, способствуют развитию тяжелой энцефалопатии и заболевания печени, кроме того, выявляются новые нарушения, возникающие в результате дефектов взаимодействия между митохондриями и их окружением в клетке.
а) Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды. Дети с MELAS могут не иметь отклонений в течение первых нескольких лет, но постепенно у них замедляется моторное и когнитивное развитие и становится очевидным низкий рост. Клинический синдром характеризуется:
1) рецидивирующими инсультоподобными эпизодами гемипареза/др. очаговыми неврологическими симптомами с патологией, свидетельствующей об очаговых поражениях ГМ по данным КТ/МРТ, чаще наблюдаемой в задней височной, теменной и затылочной долях;
3) по крайней мере двумя из следующих признаков: оча-говые/генерализованные судороги, слабоумие, повторяющиеся мигрени и рвота.
По данным одного наблюдения, заболевание дебютировало <15 лет у 62% пациентов, гемианопсия/кортикальная слепота были наиболее распространенными его проявлениями. Часто отмечается повышение белка в СМЖ. Наиболее распространенной мутацией для возникновения данной патологии является мутация MELAS 3243 на мтДНК, которая также может вызывать разл. комбинации непереносимости ФН, миопатии, офтальмоплегии, пигментной ретинопатии, ГКМП/ДКМП, дефектов сердечной проводимости, глухоты, эндокринопатии (СД) и проксимальной дисфункции почечных канальцев. Было сообщено о ряде др. мутаций, у двух пациентов описаны двусторонние роландические поражения и эпилепсия partialis continua, обусловленные мутациями мтДНК в 10158Т >С и 10191Т >С. MELAS — это прогрессирующее заболевание, которое может встречаться у братьев и сестер.
Однако большинство родственников матерей пациентов с MELAS имеют либо слабые проявления, либо вовсе не больны. MELAS сопровождается сменой эпизодов инсульта, в результате приводящих к деменции.
Регионарная церебральная гипоперфузия м.б. обнаружена с помощью однофотонной эмиссионной КТ, а МР-спектроскопия позволяет обнаружить участки очагов молочнокислого ацидоза в ГМ. При неврологическом исследовании выявляют корковую атрофию с инфарктоподобными поражениями как в корковых, так и в подкорковых структурах, кальцификацию базальных ганглиев и дилатацию желудочков. В биоптатах мышечной ткани, как правило, обнаруживают рваные красные волокна. Митохондриальные скопления и аномалии, вызывающие митохондриальную ангиопатию, обнаружены в гладкомышечных клетках сосудов и артериол ГМ, в эпителиальных клетках и кровеносных сосудах сосудистого сплетения. Во многих случаях при биохим. анализе мышц выявляется дефицит комплекса I; однако также были задокументированы множественные дефекты с участием комплексов I, III и IV.
При клиническом подозрении на MELAS для постановки диагноза обычно используются таргетное молекулярное тестирование на конкретные мутации/анализ последовательностей и сканирование мутаций.
Поскольку количество мутантных геномов в крови ниже, чем в мышцах, мышца более предпочтительна для исследования. Наследование осуществляется по материнской линии, и существует высокоспецифичная, хотя и не исключительная, точечная мутация nt 3243 в гене tRNALeu(UUR) мтДНК у 80% пациентов. Еще 7,5% имеют точечную мутацию nt 3271 в гене tRNALeu(UUR). Третья мутация была идентифицирована в nt 3252 в гене tRNALeu(UUR). Прогноз у пациентов с развернутой клинической картиной неблагоприятный. Терапевтические исследования сообщают о некоторой эффективности ГКС, коэнзима Q10, никотинамида, карнитина, креатина, рибофлавина и разл. их комбинации; успешно применение L-аргинин, ресвератрола и нового в-ва, EPI-743, аналога коэнзима Q10 (по данным доклинических исследований).
б) Миоклоническая эпилепсия и "рваные красные волокна". Синдром миоклонической эпилепсии и рваных красных волокон характеризуется прогрессирующей миоклонической эпилепсией, митохондриальной миопатией и мозжечковой атаксией с дизартрией, и нистагмом. Дебит возможен в детстве/во взрослой жизни, течение м.б. медленно/быстро прогрессирующим. Др. симптомы включают слабоумие, нейросенсорную тугоухость, оптическую атрофию, периферическую невропатию и спастичность. Поскольку у некоторых пациентов есть нарушение глубокой чувствительности и вальгусная установка, это состояние можно спутать с атаксией Фридрейха (Friedreich). Значительное число пациентов имеют положительный семейный анамнез и невысокий рост. Это заболевание передается по материнской линии.
Патологоанат. находки включают повышенную концентрацию лактата в сыворотке крови, рваные красные волокна при биопсии мышц, выраженную потерю нейронов и глиоз, поражающие, в частности, зубчатое ядро и нижний оливарный комплекс с некоторым выпадением клеток Пуркинье (Purkinje) и нейронов красного ядра. Отмечается бледность задних столбов спинного мозга и дегенерация грацильных и клиновидных ядер. Биохимия мышц выявляет вариабельные дефекты комплекса III, комплексов II и IV, комплексов I и IV/только комплекса IV. Более 80% случаев вызваны гетероплазматической G-точечной мутацией в nt 8344 гена tRNALys мтДНК. Сообщалось о пациентах с мутацией от Т до С в гене nt 8356 tRNALys. Для диагностики данного синдрома используется таргетный мутационный анализ/мутационный анализ после секвенирования митохондриального генома.
Специфической терапии не существует, хотя коэнзим Q10 оказался эффективным у матери и дочери с мутацией синдрома миоклонической эпилепсии и рваных красных волокон. Сообщается, что противосудорожный ЛП леветирацетам при этом расстройстве снижает миоклонус и миоклонические судороги.
в) Синдром невропатии, атаксии и пигментного ретинита. Это наследственное заболевание, передающееся по материнской линии, проявляется либо синдромом Ли, либо нейрогенной слабостью и синдромом невропатии, атаксии и пигментного ретинита, а также припадками. Заболевание вызвано точечной мутацией в nt 8993 в гене субъединицы 6 АТФазы. Тяжесть заболевания, по-видимому, имеет тесную корреляцию с процентным содержанием мутантной мтДНК в лейкоцитах. У пациентов с синдромом невропатии, атаксии и пигментного ретинита наблюдаются два клинических варианта:
1) невропатия, атаксия, пигментный ретинит, деменция и атаксия;
2) тяжелая инфантильная энцефалопатия, напоминающая синдром Ли, с поражением базальных ганглиев по данным МРТ.
г) Наследственная оптическая невропатия Лебера. Наследственная оптическая невропатия Лебера обычно характеризуется дебютом острой/подострой потери зрения в 18-30 лет, вызванной тяжелой двусторонней атрофией зрительного нерва, хотя сообщалось, что данный синдром развивается и у детей >5 лет. Три мутации мтДНК составляют большинство случаев наследственной оптической невропатии Лебера, и по крайней мере 85% пациентов — молодые мужчины. Х-сцепленный фактор может модулировать экспрессию точечной мутации мтДНК. Классические офтальмологические признаки включают циркумпапиллярную телеангиэктатическую микроангиопатию и псевдоотек диска зрительного нерва. Переменные признаки могут включать мозжечковую атаксию, гиперрефлексию, симптом Бабинского, психиатрические проблемы, периферическую невропатию и нарушения сердечной проводимости (синдром предвозбуждения).
Некоторые случаи вызваны значительными распространенными поражениями белого в-ва, как это наблюдается при рассеянном склерозе. Молочнокислый ацидоз и рваные красные волокна при наследственной оптической нейропатии Лебера, как правило, отсутствуют. Описано >11 точечных мутаций мтДНК, включая гомоплазматический G-переход в nt 11 778 гена субъединицы ND4 комплекса I. Последняя мутация приводит к замене остатка аргинина на гистидин в 340-й аминокислоте и составляет 50-70% случаев в Европе и >90% случаев в Японии. Происхождение некоторых типов наследственной оптической нейропатии Лебера вызвано др. точечными мутациями, обуславливающими развитие сложных неврологических расстройств, они могут иметь общие черты с синдромом MELAS и с детским двусторонним стриатальным некрозом. В одной семье было описано раннее начало с прогрессирующей генерализованной дистонией с двусторонним стриатальным некрозом, вызванными гомоплазматической мутацией G14459А в гене ND6 мтДНК, которая может провоцировать развитие наследственной оптической нейропатии Лебера с дистонией или без нее.
Идебенон и EPI-743 показали свою эффективность при лечении этого расстройства.
д) Синдром Кэрнза-Сейра. Синдром Кэрнза-Сейра — это характерное полиорганное расстройство, включающее внешнюю офтальмоплегию, блокаду сердца и пигментный ретинит с началом <20 лет, вызванное единичными делециями мтДНК. Также должно быть по крайней мере один из перечисленных признаков: блокада сердца, мозжечковый синдром, повышение уровня белка в СМЖ >100 мг/дл. Др. неспецифические, но общие признаки включают слабоумие, нейросенсорную тугоухость и множественные эндокринные нарушения, включая низкий рост, СД и гипопаратиреоз. Прогнозирование осуществляется с осторожностью, несмотря на установку кардиостимулятора, и постепенно ухудшается, смерть наступает в третьем/четвертом десятилетии. Необычные клинические проявления могут включать ацидоз почечных канальцев и синдром Лоу (Lowe). Есть также данные о сочетании у некоторых детей синдрома Кэрнза-Сейра с инсультоподобными эпизодами.
Биопсия мышц демонстрирует рваные красные волокна и вариабельные цитохром-С-оксидаза-«-» волокна. У большинства пациентов есть делеции мтДНК, а у некоторых — дупликации. Это м.б. новые мутации, объясняющие в целом спорадический характер синдрома. В нескольких семьях отмечена АуД-передача.
Пациенты должны находиться под пристальным наблюдением на предмет эндокринных нарушений, которые необходимо лечить. Сообщается, что коэнзим Q10 в данном случае обладает некоторым «+» эффектом; сообщалось о эффективности применения фолиевой кислоты при низком уровне фолата. Известно также о «+» влиянии кохлеарного имплантата при глухоте.
Спорадическая прогрессирующая наружная офтальмоплегия с рваными красными волокнами — это клинически доброкачественное состояние, характеризующееся подростковой/юношеской офтальмоплегией, птозом и слабостью проксимального пояса конечностей. Она медленно прогрессирует и совместима с относительно нормальной жизнедеятельностью. В мышечных биоптатах рваные красные волокна и цитохром-С-оксидаза-«-» волокна. У 50% пациентов с прогрессирующей наружной офтальмоплегией есть делеции мтДНК и нет семейного анамнеза.
е) Обратимая инфантильная миопатия с недостаточностью цитохром-С-оксидазы. Мутации в мтДНК ответственны за обратимую форму тяжелой нервно-мышечной слабости и гипотонии у младенцев, которая является результатом унаследованной от матери гомоплазматической мутации т.14674Т>С mt-tRNAGlu, вызванной дефицитом цитохром-С-оксидазы. Дети в течение первых нескольких недель жизни страдают гипотонией, выраженной мышечной слабостью и имеют очень высокий уровень лактата в сыворотке крови, таким детям часто требуется проведение ИВЛ. Однако на питание и психомоторное развитие это не влияет. В мышечных биоптатах, взятых в неонатальном периоде, обнаруживаются рваные красные волокна и недостаточная активность цитохром-С-оксидазы, но в течение 5-20 мес они исчезают, и дети спонтанно выздоравливают. До улучшения состояния детей с данной патологией трудно отличить от детей с летальными митохондриальными нарушениями. Механизм этого восстановления не установлен, но он может отражать эволюционный сдвиг в митохондриальных РНК позже в младенчестве.
Это обратимое расстройство наблюдается только при дефиците цитохром-С-оксидазы, вызванном мутацией 14674Т>С mttRNAGlu, поэтому предполагается, что младенцы с этим типом тяжелой мышечной гипотонии в неонатальном периоде должны быть обследованы на эту мутацию с целью определения прогноза.
ж) Болезнь Ли (подострая некротизирующая энцефаломиопатия). Болезнь Ли — прогрессирующее дегенеративное расстройство, проявляющееся в младенчестве проблемами с кормлением и глотанием, рвотой и задержкой развития, сопровождающимися молочнокислым ацидозом и поражениями ствола ГМ и/или базальных ганглиев по данным МРТ (табл. 5). Существует несколько генетически детерминированных причин болезни Ли, которые являются результатом мутаций ядерной ДНК в генах, кодирующих компоненты дыхательной цепи: дефицит комплекса пируватдегидрогеназы, дефицит комплекса I или II, дефицит комплекса IV, дефицит комплекса V (АТФазы) и дефицит кофермента Q10. Эти дефекты могут возникать споради-чески/наследоваться АуР-путем, как в случае дефицита цитохром-С-оксидазы; Х-сцепленным путем, как в случае дефицита пируватдегидрогеназы Е1а; и материнской передачей, как в случае дефицита комплекса V (мутация АТФазы 6 nt 8993). Примерно 30% случаев вызваны мутациями в мтДНК.
Замедление этапов двигательного, речевого развития и генерализованные судороги, слабость, гипотония, атаксия, тремор, пирамидные признаки и нистагм являются важными симптомами этого синдрома. Прерывистое дыхание с сопутствующими вздохами/всхлипываниями также характерны и предполагает дисфункцию ствола ГМ. У некоторых пациентов наблюдается наружная офтальмоплегия, птоз, пигментный ретинит, атрофия зрительного нерва и снижение остроты зрения. Аномальные результаты на КТ/МРТ представлены билатерально симметричными областями пониженного сигнала в базальных ганглиях и стволе ГМ, а также повышенным содержанием молочной кислоты на MP-спектроскопии (рис. ниже). Патологические изменения состоят из очаговых симметричных участков некроза в таламусе, базальных ганглиях, тегментальном сером в-ве, перивентрикулярных и периакведуктальных областях ствола ГМ и задних столбах спинного мозга. Микроскопически это губчатые очаги с кистозной кавитацией с потерей нейронов, демиелинизацией и пролиферацией сосудов. Характерны повышенные уровни лактата в сыворотке крови и ликворе, а также ГКМП, печеночная недостаточность и дисфункция почечных канальцев.
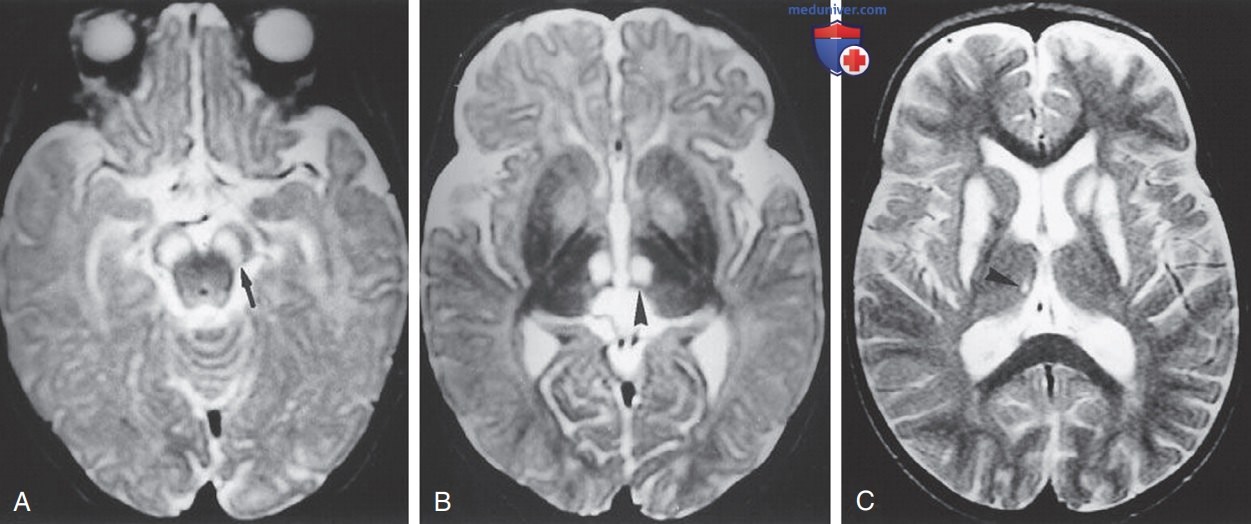
Синдром Ли. Осевая Т2-взвешенная магнитно-резонансная томограмма (TR/TE/NEX =3,000/120/1 мс) девочки 8 мес с синдромом Ли вследствие мутации SURF1 демонстрирует гиперинтенсивные поражения в черной субстанции и медиальных ядрах таламуса (A, B). Последующие изображения (TR/TE/NEX =2,028/120/2 мс) в возрасте 26 мес (C) демонстрируют гиперинтенсивность путамена и левого хвостатого ядра.
Общий прогноз неблагоприятен, но у некоторых пациентов наблюдаются длительные периоды ремиссии. Основного лечения заболевания не существует, но ряд витаминов, включая рибофлавин, тиамин и коэнзим Q10, часто назначают с целью улучшения функций митохондрий. Также применялись биотин, креатин, сукцинат, идебенон и EPI-743, диета с высоким содержанием жиров. Фенобарбитал и вальпроевую кислоту следует избегать из-за их ингибирующего действия на митохондриальную дыхательную цепь.
з) Синдром истощения митихондриальной ДНК. Синдром истощения митихондриальной ДНК (мтДНК) — группа АуР-расстройств, которые вызывают значительное снижение содержания мтДНК в мышцах, печени и ГМ (табл. 6). Это состояние обычно приводит к летальному исходу в младенчестве, хотя некоторые дети доживают до подросткового возраста.
и) Синдром Рейе. Энцефалопатия, ставшая редкостью, сопровождается развитием патологических изменений, характеризующихся жировой дегенерацией внутренних органов (микровезикулярный стеатоз), митохондриальными аномалиями и биохим. особенностями, отражающими нарушение митохондриального метаболизма.
Периодический Рейе-подобный синдром встречается у детей с генетическими дефектами окисления жирных кислот, напр. недосточность плазмалеммаль-транспортера карнитина, карнитин-пальмитоилтрансферазы I и II, карнитин-ацилкарнитинтранслоказы, средне-/длинноцепочечных ацил-коэнзим А дегидрогеназы, множественной ацил-коэнзим А дегидрогеназы, длинных цепочек с L-3 гидроксиацил-коэнзим А дегидрогеназы, трифункционального белка. Эти нарушения проявляются рецидивирующей гипогликемической и гипокетотической энцефалопатией и наследуются по АуР-типу. Др. потенциальные врожденные нарушения метаболизма, характеризующие синдром Рейе, включают дефекты цикла мочевины (орнитинтранскарбамилаза, карбамилфосфатсинтетаза) и некоторые органические ацидурии (глутаровая ацидурия типа I), дефекты дыхательной цепи и дефекты углеводного обмена (непереносимость фруктозы).