Дистония — расстройство движения, характеризующееся длительными мышечными сокращениями, часто вызывающими скручивание/повторяющиеся движения/ненормальные позы. Основными причинами дистонии являются первичная генерализованная дистония, ЛП, нарушения обмена в-в и перинатальная асфиксия (табл. 2 и 13).
а) Наследственные первичные дистонии. Первичная генерализованная дистония, называемая первичной торсионной дистонией/dystonia musculorum deformans, обусловлена группой генетических нарушений с дебютом в детском возрасте (рис. 1). Одна из форм, которая чаще встречается в еврейской популяции ашкенази, вызвана доминантной мутацией в гене DYT1, кодирующем АТФ-связывающий белок торсина. Начальным проявлением дистонии DYT1 часто является прерывистое одностороннее положение нижней конечности с ее выпрямлением и ротацией.
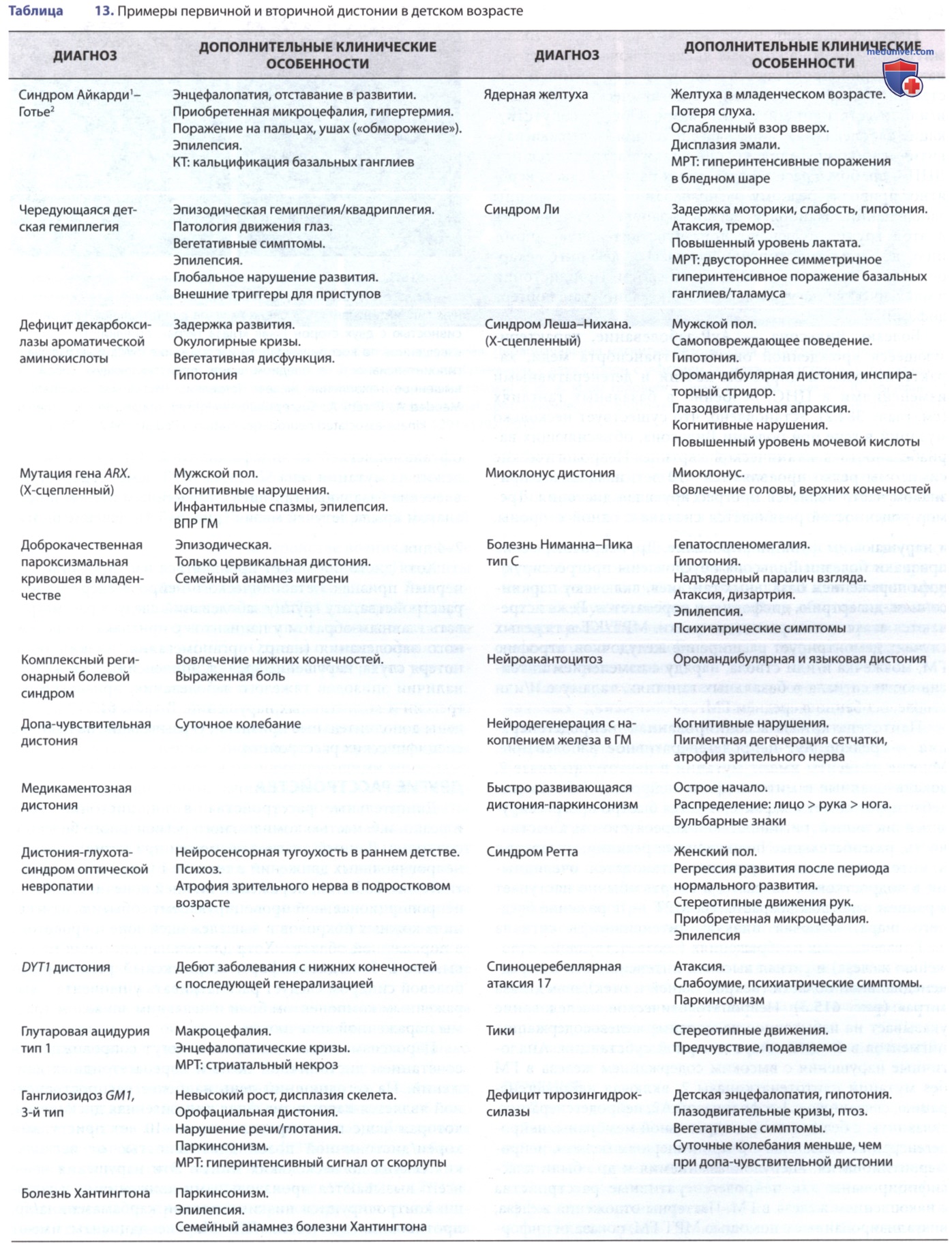
Рисунок 1. Синдромы с дистонией в качестве основного/преобладающего признака; перечислены варианты первичной дистонии/дистонии + синдромы, которые обычно дебютируют с ее появления. Наиболее распространенная локализация дистонии обозначена на гомункуле красным цветом, менее распространенные — розовым. Распределение по возрасту начала заболевания показано синей полосой, средний возраст — синим ромбом, а редкие, но зафиксированные крайние отклонения — внелинейными синими черточками. Типичные темпы прогрессии и вероятность генерализации обозначены желтыми графиками. Обратите внимание, что гомункулы и графики представляют собой наиболее распространенные клинические проявления, но не редки вариации по этим осям
В конечном счете в дистонию могут вовлекаться все четыре конечности, и осевая мускулатура/оставаться локализованной на одной конечности. Вовлечение головы при дистонии DYT1 возможно, но реже по сравнению с дистонией без DYT1. Существует широкий клинический спектр, варьирующий даже внутри семей.
Если в семейном анамнезе дистония отсутствует, все равно следует рассматривать этот диагноз, учитывая в/семейную вариабельность клинической картины.
Было идентифицировано >10 локусов для генов торсионной дистонии (DYT1-DYT24). Одним из них является АуД-расстройство, обуславливающее развитие дистонии (допа-чувствительная дистония, DYT5a), называемой синдромом Сегавы (Segawa). Ген допа-чувствительной дистонии кодирует гуанозинтрифосфатциклогидролазу 1, фермент, ограничивающий скорость синтеза тетрагидробиоптерина, который является кофактором синтеза нейромедиаторов дофамина и серотонина.
Т.о., генетическая мутация приводит к дефициту дофамина. Отличительной чертой этого расстройства, особенно у подростков и взрослых, является суточная вариабельность: симптомы усугубляются в течение дня и могут временно улучшаться после сна. Пациенты с ранним дебютом, имеющие замедленную/измененную походку вследствие дистонии нижних конечностей, м.б. легко спутаны с пациентами с дистонической формой ДЦП. Следует отметить, что при наличии прогрессирующей дистонии, суточных колебаний и утраты ранее приобретенных двигательных навыков предварительный диагноз ДЦП должен быть пересмотрен.
Допа-чувствительная дистония имеет хороший терапевтический ответ на небольшие суточные дозы леводопы. Чувствительность к леводопе важна даже при поздней постановке диагноза, до развития контрактур. Реже АуР-форма этого расстройства вызывается мутациями в гене тирозингидроксилазы.
Миоклонус-дистония (DYT11), вызванная мутациями в гене эпсилонсаркогликана, характеризуется дистонией, вовлекающей верхние конечности, голову и/или шею, миоклоническими движениями в этих областях. Несмотря на то что миоклонус и дистония часто сочетаются, каждое из них сущестовать изолированно. Повторный миоклонус может принимать тремороподобный вид, что носит название дистонического тремора. Уменьшение симптомов после приема алкоголя, о чем сообщают взрослые члены семьи с данной патологией, может быть важным ключом к этому диагнозу.
Общим для наследственных дистоний является значительная в/семейная вариабельность клинических проявлений, распределения и тяжести. При первичных дистониях, хотя основными клиническими признаками являются двигательные нарушения, м.б. повышен риск развития тяжелой депрессии. Тревожность, ОКР и депрессия были зарегистрированы при синдроме миоклонус-дистонии. Не следует упускать из виду скрининг на сопутствующие психические заболевания представителей этой популяции.
б) Лекарственные дистонии. Некоторые ЛП способны вызывать непроизвольные движения/двигательные расстройства у детей и взрослых. Блокаторы дофамина, включая антипсихотики (напр., галоперидол) и противорвотные ЛП (напр., метоклопрамид, прохлорперазин), а также атипичные антипсихотики (напр., рисперидон, арипипразол), могут вызывать острые дистонические реакции/замедленные (запоздалые) двигательные расстройства.
Острые дистонические реакции, возникающие в первые дни применения, как правило, затрагивают лицо/шею и проявляются в виде кривошеи, ретроколлиса, окулогирического криза и выпячивания языка. Могут возникать угрожающие жизни проявления в виде ларингоспазма и нарушения проходимости ДП, требующие оперативной реакции и лечения. В/в введение дифенгидрамина 1-2 мг/кг (максД 50 мг) может быстро купировать лекарственную дистонию. Степень потенции блокатора дофамина, молодой возраст и предшествующие дистонические реакции могут быть предрасполагающими факторами. Острые дистонические реакции также описаны при применении цетиризина.
Выраженная ригидность в сочетании с высокой ТТ, вегетативными симптомами (тахикардия, потоотделение), делирием и дистонией являются признаками злокачественного нейролептического синдрома, который обычно возникает через несколько дней после начала/увеличения дозы нейролептического/отмены дофаминергического ЛП. В отличие от острых дистонических реакций, которые протекают в течение нескольких дней, нейролептический злокачественный синдром обычно возникает в течение месяца после начала приема ЛП/увеличения дозы.
Замедленные непроизвольные движения, поздние дискинезии развиваются в условиях хронического применения нейролептиков обычно >3 мес. Характерно вовлечение лица, особенно рта, губ и/или челюсти, при жевании/ высовывании языка. Риск развития поздней дискинезии, которая значительно реже встречается у детей по сравнению со взрослыми, возрастает по мере увеличения дозы ЛП, продолжительности лечения и полипрагмазии.
Имеются данные, позволяющие предположить, что дети с РАС также м.б. подвержены риску развития этого лекарственно-индуцированного двигательного расстройства. В отличие от острых дистонических реакций и нейролептического злокачественного синдрома, прекращение приема ЛП может не привести к клиническому улучшению. У этих пациентов может оказаться эффективным применение дофамин-деплеторов, таких как резерпин/тетрабеназин.
Терапевтические дозы фенитоина, карбамазепина и вальпроата редко вызывают прогрессирующую дистонию у детей с эпилепсией, особенно имеющих структурную аномалию ГМ.
При обследовании впервые возникшей дистонии крайне важно тщательно изучить историю назначений и потенциальное воздействие ЛП.
в) Церебральный паралич. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
г) Нарушение обмена веществ. Нарушения метаболизма моноаминовых нейротрансмиттеров, одним из которых является допа-чувствительная дистония, проявляются в младенчестве и раннем детстве дистонией, гипотонией, окулогирическими кризами и/или вегетативными симптомами. Общие сопутствующие заболевания, такие как эпилепсия, задержка развития и микроцефалия, которые также встречаются при ДЦП и др. более распространенных расстройствах, вероятно, приводят редкому распознаванию данной группы заболеваний.
Наиболее распространенные нарушения в этой группе включают допа-чувствительную дистонию, дефицит тирозингидроксилазы и дефицит декарбоксилазы ароматической аминокислоты. При дистонии в младенчестве могут выявляться аномалии транспортера дофамина.
Болезнь Вильсона — АуР-заболевание, характеризующееся врожденной ошибкой транспорта меди, характеризующееся циррозом печени и дегенеративными изменениями в ЦНС, особенно в базальных ганглиях. Установлено, что существует несколько мутаций в гене при болезни Вильсона, объясняющих вариабельность в клинической картине. Неврологические симптомы редко проявляются <10 лет, начальным признаком часто является прогрессирующая дистония.
Тремор конечностей развивается сначала с одной стороны, но со временем он становится грубым, генерализованным и нарушающим функционирование.
Др. неврологические признаки болезни Вильсона обусловлены прогрессирующим поражением базальных ганглиев, включают паркинсонизм, дизартрию, дисфонию и хореоатетоз. Реже встречаются атаксия и пирамидные знаки. МРТ/КТ в тяжелых случаях демонтрирует расширение желудочков, атрофию ГМ, мозжечка и/или ствола, наряду с изменением интенсивности сигнала в базальных ганглиях, таламусе и/или стволе, особенно в среднем ГМ.
Пантотенаткиназа-ассоциированная нейродегенерация — редкое АуР-нейродегенеративное заболевание. Многие пациенты имеют мутации в пантотенаткиназе 2, локализованные в митохондриях нейронов. Заболевание дебютирует <6 лет и характеризуется быстро прогрессирующей дистонией, ригидностью и хореоатетозом. Спастичность, разгибательные подошвенные реакции, дизартрия и интеллектуальное нарушения становятся очевидными в подростковом возрасте, и смерть обычно наступает в раннем взрослом возрасте.
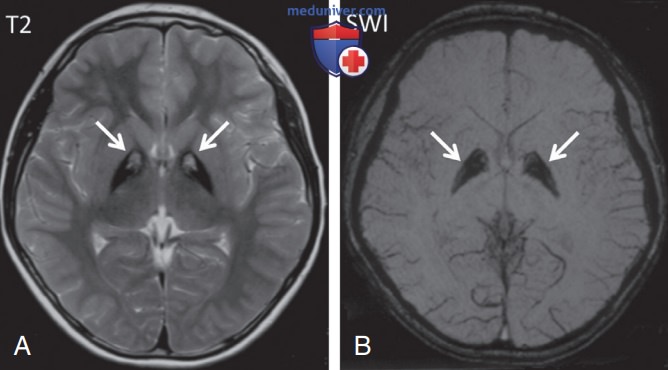
На МРТ — поражение бледного шара, включая низкую интенсивность сигнала на Т2-взвешенных изображениях (соответствующих отложению железа) и сигнал высокой интенсивности в переднемедиальной области (некроз тканей и отек)/знак «глаза тигра» (рис. 2). Невропатологическое исследование указывает на избыточное накопление железосодержащих пигментов в бледном шаре и черной субстанции.
Рисунок 2. Пантотенаткиназа-ассоциированная нейродегенерация: A — осевое Т2-взвешенное изображение, показывающее симметричную гипоинтенсивность в бледном шаре с центральной гиперинтенсивностью с двух сторон (знак «глаза тигра», стрелки); B — осевое взвешенное по восприимчивости изображение (SWI), показывающее гипоинтенсивность в бледном шаре, представляющую собой повышенное накопление железа (стрелки)
Аналогичные нарушения с высоким содержанием железа в ГМ без мутаций пантотенаткиназы 2, включая нейродегенерацию, связанную с фосфолипазой А2, нейродегенерацию, связанную с белком митохондриальной мембраны, нейродегенерация, связанная с β-пропеллерным белком, нейроферритинопатия, ацерулоплазминемия и др., были классифицированы как нейродегенеративные расстройства с накоплением железа в ГМ. Паттерны отложения железа, визуализированные с помощью МРТ ГМ, показали информативность данного метода для ДД этих расстройств.
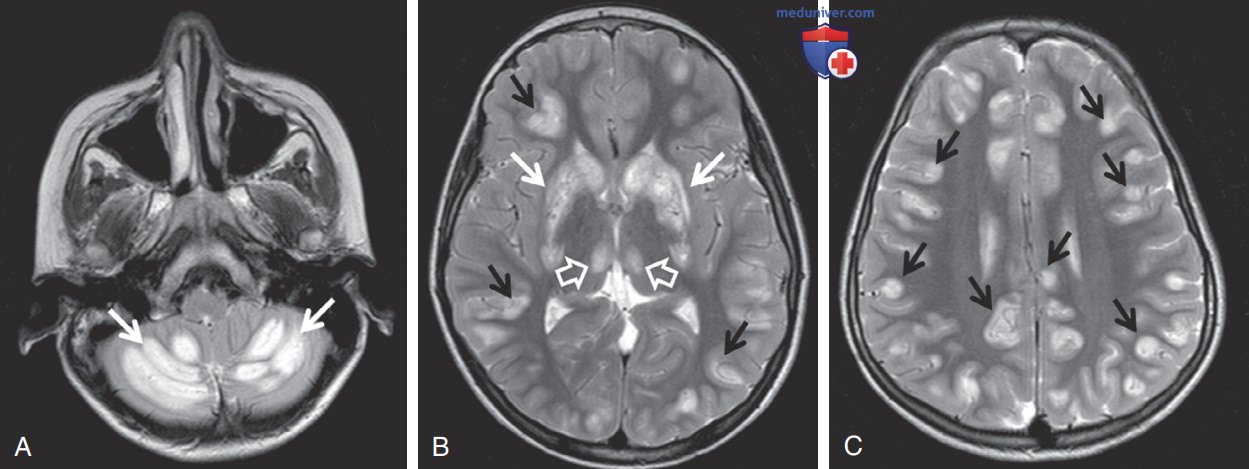
Чувствительное к биотину поражение базальных ганглиев проявляется эпизодами острой дистонии, внешней офтальмоплегией и энцефалопатией. Ответственность лежит на мутации гена SLC19A3. МРТ демонстрирует вовлечение базальных ганглиев с вазогенным отеком и признаком крыла летучей мыши (рис. 3). Лечение биотином и тиамином приводит к улучшению состояния через 2-4 дня.
Рисунок 3. Чувствительное к биотину поражение базальных ганглиев. Первоначальная магнитно-резонансная томограмма головного мозга показала повышение интенсивности сигнала на Т2-взвешенных изображениях с двух сторон с участием (А) мозжечка (стрелки), (В) базальных ганглиев (белые стрелки), медиального ядра таламуса (открытые стрелки) и (B, C) коры головного мозга (черные стрелки)
Хотя дистония может проявляться изолированно как первый признак метаболического/нейродегенеративного расстройства, эту группу заболеваний следует рассматривать главным образом у пациентов с признаками системного заболевания (напр., органомегалия, низкий рост, потеря слуха, нарушение зрения, эпилепсия), а также при наличии эпизодов тяжелого заболевания, признаков регрессии и когнитивных нарушений. В табл. 12 приведены дополнительные признаки, указывающие на наличие специфических расстройств.
д) Другие расстройства. Двигательные расстройства, в т.ч. дистония, хоть и редко, м.б. частью комплексного регионарного болевого синдрома. Данный диагноз возможен при возникновении непроизвольных движений в течение 1 года после травматического события, поражении нижней конечности, боли, непропорциональной провоцирующему событию, изменениях кожных покровов в вышележащей зоне и кровотока в пораженной области.
Хотя длительная дистония может вызывать боль/дискомфорт, комплексный регионарный болевой синдром следует рассматривать у пациента с выраженным компонентом боли и недавним эпизодом травмы пораженной конечности.
Пароксизмальные дискинезии могут сопровождаться сочетанием дистоничной позы и хореоатетоидных движений. На сегодняшний день наиболее распространенной является пароксизмальная кинезигенная дискинезия, которая чаще всего проявляется в ~10 лет приступами хореи/дистоничной позой длительностью от нескольких секунд до нескольких минут. Эти нарушения чаще всего вызываются произвольными движениями и хорошо контролируются низкими дозами карбамазепина/др. противоэпилептических ЛП. Многие пациенты имеют мутацию в PRRT2, трансмембранном белке, который взаимодействует с SNAP25.
Пароксизмальная некинезигенная дискинезия характеризуется длительными приступами, вызванными эмоциональным стрессом/алкоголем. Приступы случаются реже, возможно, несколько раз в год, но могут длиться часами. Эта форма дискинезии менее чувствительна к лечению. Наконец, самой редкой формой пароксизмальной дискинезии является дистония, вызванная ФН. Дистония при этом расстройстве возникает после периодов длительной ФН и длится 10-30 мин. Пациенты также могут страдать от мигрени и эпилепсии. Это расстройство вызвано мутациями в SLC2A1, который кодирует белок-транспортер глюкозы типа 1, и является частью синдрома дефицита GLUT-1.
Отчеты о таких случаях показывают, что у некоторых пациентов отмечается улучшение на фоне кетогенной диеты.
Существуют расстройства, присущие исключительно детскому возрасту, требующие упоминания в данной статье. Доброкачественная инфантильная пароксизмальная кривошея характеризуется рецидивирующими эпизодами цервикальной дистонии, начинающимися в первые месяцы жизни. От одного эпизода к другому могут чередоваться стороны, возможно сохранение симптоматики во время сна. Сопутствующие признаки и симптомы включают раздражительность, бледность, рвоту, головокружение, атаксию и иногда дистонию конечностей. В семейном анамнезе часто отмечается мигрень и/или морская болезнь у родственников первой степени.
Несмотря на высокую частоту приступов, при визуализации выявляется норма, прогноз благоприятный, постепенное улучшение с разрешением к 3 годам.
При альтернирующей гемиплегии детского возраста характерной чертой расстройства является эпизодическая гемиплегия, поражающая обе стороны тела. Однако пациенты также страдают от эпизодов дистонии, которые могут длиться от нескольких минут до нескольких дней. В среднем оба расстройства начинаются в ~6 мес. Эпизодические аномальные движения глаз наблюдаются у значительной части пациентов (93%) с началом заболевания уже на первой неделе жизни. Альтернирующая гемиплегия вызвана мутациями в генах АТР1А2 и АТР1A3. Патологические проявления провоцируются колебаниями температуры, определенными продуктами питания и воздействием воды.
Со временем появляются эпилепсия и когнитивные нарушения, а непроизвольные движения из эпизодических трансформируются в постоянные. Дебют в младенчестве и пароксизмальная природа симптомов на ранних стадиях заболевания являются ключевыми признаками этого диагноза.
Еще одно расстройство, связанное с мутациями в АТР1А3, — быстро прогрессирующий дистонический паркинсонизм, часто встречается у подростков с развитием острой/подострой прогрессирующей дистонии и брадикинезии, часто после стресса, напр. перенесенного заболевания. Хотя классические формы этих двух последних расстройств обычно вызываются неперекрывающимися мутациями, молекулярная генетика позволила идентифицировать пациентов с промежуточными фенотипами.
Наконец, хотя это диагноз исключения, наличие аномальных движений/частичной инвалидизации может указывать на психогенную дистонию у детей старшего возраста. Существует значительное совпадение в особенностях органических и психогенных двигательных расстройств, что затрудняет постановку диагноза. Напр., как органические, так и психогенные двигательные расстройства могут усугубляться в условиях стресса и нивелироваться при расслаблении/во сне. История болезни должна включать обзор недавних стрессоров, психиатрических симптомов и контактов с др. людьми с аналогичными расстройствами.
При обследовании меняющееся двигательное расстройство, непоследовательный моторный/сенсорный ответ, реакция на внушение выступают в пользу психогенной природы двигательного расстройства. Ранняя диагностика этой патологии может снизить заболеваемость, вызванную ненужными диагностическими и интервенционными процедурами.
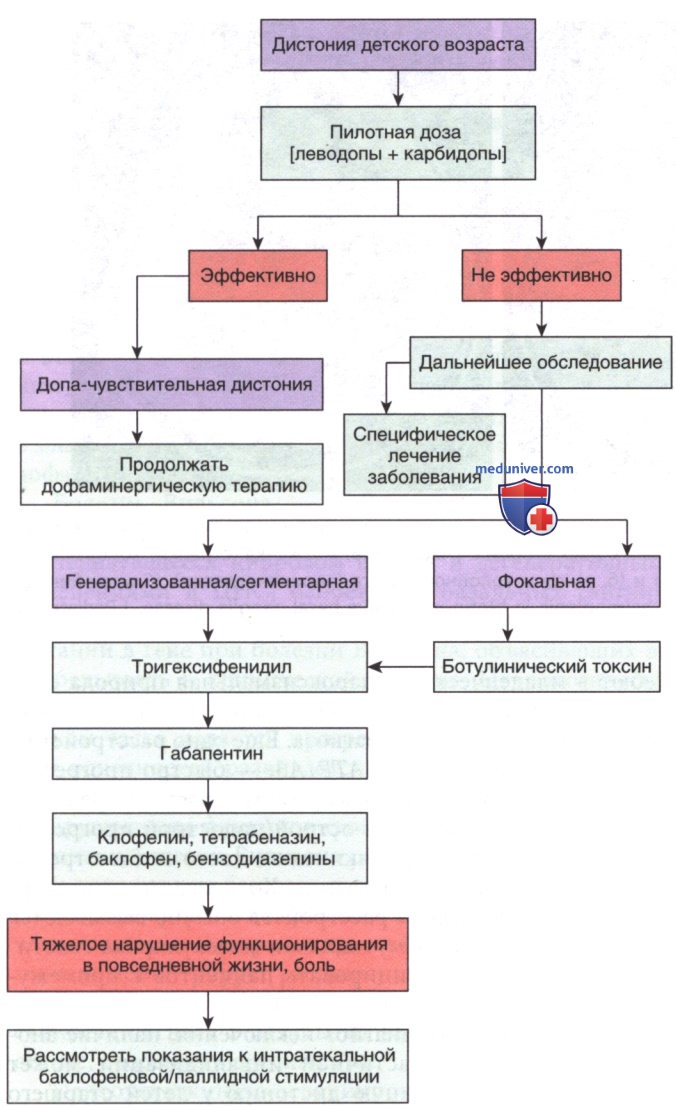
Диагностика представлена в табл. 14 и на рис. 4.
Рисунок 4. Терапевтические подходы к лечению детской дистонии. Лекарственный препарат следует применять по возможности в низких дозах. Высокие дозы и полипрагмазия неизбежны в случае тяжелых форм дистонии, вызывающих боль и мешающих ежедневному функционированию, комфорту в положении сидя и во время сна. Как и в случае с эпилепсией, трудно поддающейся терапии, следует рассмотреть вопрос о функциональной нейрохирургии, если >2 лекарственных препаратов неэффективны.
е) Лечение. Дети с генерализованной дистонией, в т.ч. с вовлечением мышц, участвующих в глотании, могут отвечать на терапию антихолинергическим ЛП тригексифенидил. Титрование происходит медленно в течение нескольких месяцев в попытке уменьшить нежелательные побочные эффекты, напр. задержку мочи, спутанность сознания и нарушение зрения. Дополнительными ЛП, показавшими свою эффективность, являются леводопа и диазепам. Сегментарная дистония, напр., кривошея, часто поддается лечению инъекциями ботулотоксина.
У некоторых пациентов м.б. эффективен интратекальный баклофен, доставляемый через имплантируемый постоянный инфузионный насос. Глубокая стимуляция ГМ электродами, имплантированными в бледный шар, наиболее эффективна у детей с тяжелой первичной генерализованной дистонией. Глубокая стимуляция ГМ возможна у детей с вторичными дистониями, напр. ДЦП.
В случае медикаментозной дистонии обычно достаточно отмены ЛП и в/в введения дифенгидрамина. При злокачественном нейролептическом синдроме м.б. показан дантролен.