Нейрональный цероидный липофусциноз у ребенка - кратко с точки зрения педиатрии
Нейрональные цероидные липофусцинозы представляют собой группу наследственных нейродегенеративных лизосомальных нарушений памяти, характеризующихся потерей зрения, прогрессирующей деменцией, судорогами, снижением двигательной активности и ранней смертью.
Нейрональные цероидные липофусцинозы названы так из-за в/клеточного накопления флуоресцентных липопигментов цероида и липофусцина. Они представляют собой генетически и фенотипически гетерогенную группу расстройств (в настоящее время существует по меньшей мере девять типов заболевания), которые традиционно подразделяются по возрасту дебюта и др. клиническим признакам.
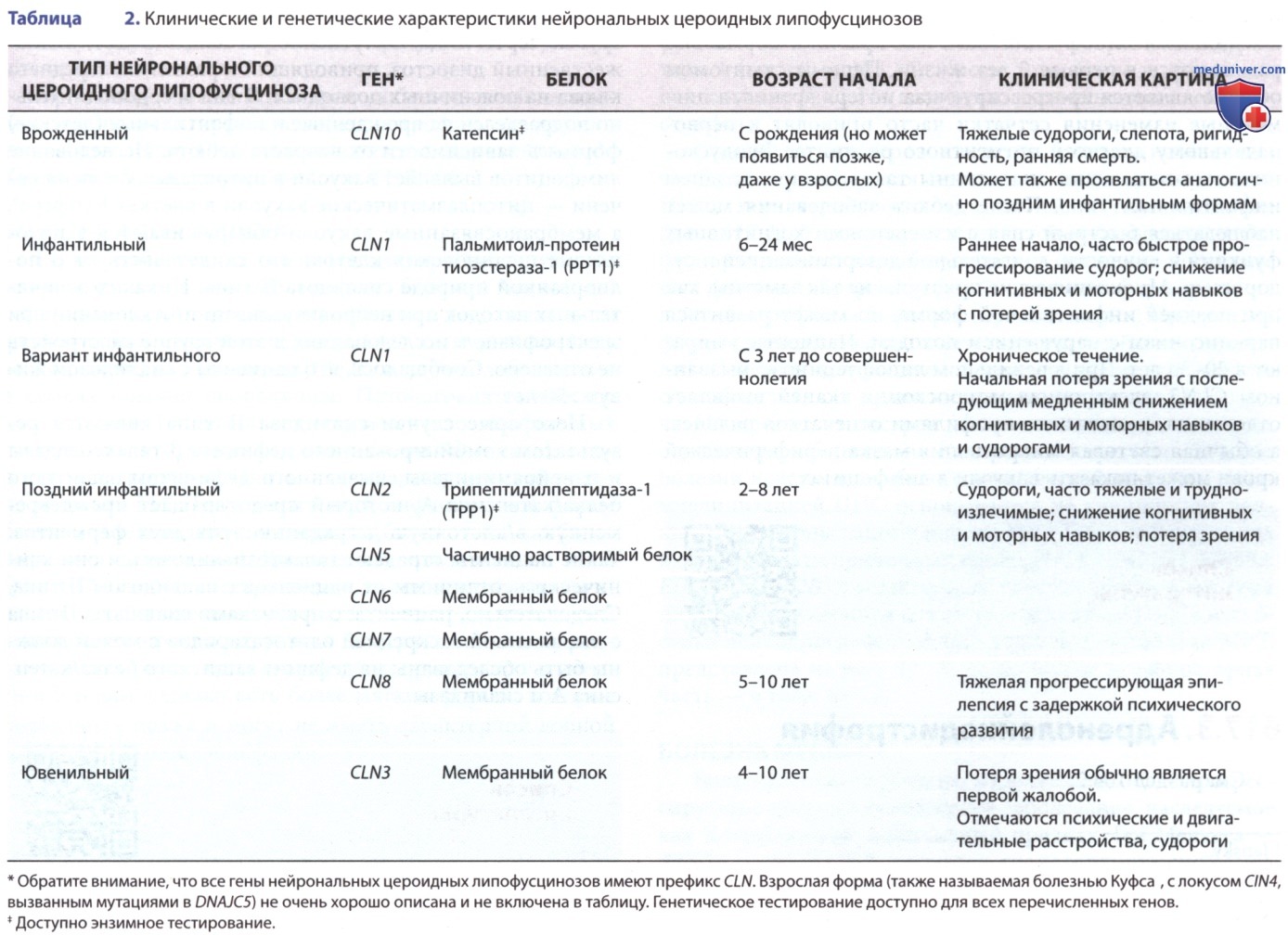
Типы отличаются друг от друга соответствующими ультраструктурными рисунками включений, наблюдаемыми при электронной микроскопии. Биопсия нейронов (ГМ, прямой кишки, конъюнктивы и кожи) ранее была необходима для постановки диагноза. С появлением ферментативных и молекулярных методов тестирования клиницисты могут определять специфические типы нейрональных цероидных липофусцинозов, используя менее инвазивные методы (табл. 2).
Нейрональный цероидный липофусциноз инфантильного типа (Халтиа-Сантувиори (Haltia, Santavuori)) дебютирует на первом году жизни с миоклонических припадков, снижения интеллекта и слепоты. При осмотре сетчатки видны атрофия зрительного нерва и коричневатое обесцвечивание макулы; характерна выраженная мозжечковая атаксия. Электроретинограмма обычно показывает малую амплитуду/отсутствие сигналов.
Смерть наступает в детстве. Инфантильная форма обусловлена АуР-мутациями гена лизосомального фермента пальмитоил-протеина тиоэстеразы-1 (РРТГ) на хромосоме 1р32. В ряде типов клеток у пациентов с нейрональным цероидным липофусцинозом отмечаются характерные в/клеточные мелкозернистые осмиофильные отложения, различимые при электронной микроскопии.
Подгруппа детей с дефицитом фермента РРТ1 имеет гораздо менее тяжелое течение с клиническими признаками, напоминающими таковые у пациентов с ювенильным началом липофусциноза. Клинически пациенты с этим вариантом имеют течение, которое часто совершенно отличается от типичной, классической, быстро дегенерирующей инфантильной формы, но при этом с соответствующим дефицитом РРТ1 и гранулярными осмиофильными отложениями внутри клеток. Нет четкого генотипа CLN1, который предсказывал бы тяжесть фенотипа.
Поздний инфантильный нейрональный цероидный липофусциноз (Янского-Билыповского (Jansky, Bielschowsky)) обычно проявляется миоклоническими приступами, начинающимися в 2-4 года у ранее здорового ребенка. Слабоумие и атаксия сочетаются с прогрессирующей потерей остроты зрения и микроцефалией. Исследование сетчатки показывает выраженное изменение сосудов, периферические пигментные аномалии черных костных спикул, атрофию зрительного нерва и тонкий коричневый пигмент в макулярной области.
Электроретинограмма и вызванные зрительные потенциалы на ранних стадиях заболевания имеют патологическую структуру. Аутофлуоресцентный материал откладывается в нейронах, фибробластах и секреторных клетках. Электронно-микроскопическое исследование биоптата кожи/конъюнктивы обычно выявляет криволинейные профили.
Данная форма заболевания м.б. вызвана АуР-мутациями нескольких разл. генов: гена CLN2, кодирующего трипептидилпептидазу-1 (TPP1), которая необходима для деградации холецистокинина-8, а также генов CLN5, CLN6 и CLN8, кодирующих мембранные белки, но они не полностью описаны. CLN8 также известен как локус синдрома Северной эпилепсии, который часто называют прогрессирующей эпилепсией с когнитивными нарушениями.
Нейрональный цероидный липофусциноз ювенильного типа (болезнь Шпильмейера-Фогта (Spielmeyer, Vogt) или Баттена (Batten)) является наиболее распространенной формой данной группы заболеваний и обычно вызывается АуР-мутациями в CLN3. (Пациентам с клиническими проявлениями ювенильной формой липофусциноза, но при этом дефицитом РРТ1/ТРР1, ставится диагноз инфантильного/позднего инфантильного типа соответственно.)
Дети, страдающие ювенильным липофусцинозом, как правило, нормально развиваются в первые 5 лет жизни. Первым симптомом обычно является прогрессирующая потеря зрения, а пигментные изменения сетчатки часто приводят к первоначальному диагнозу пигментного ретинита. Фундускопические изменения аналогичны таковым при позднем инфантильном типе. После дебюта заболевания может наблюдаться быстрый спад с изменениями когнитивных функций и личности, двигательной дезорганизацией и судорогами.
Миоклонические приступы не так заметны, как при поздней инфантильной форме, но может развиться паркинсонизм с нарушением походки. Пациенты умирают в 20-30 лет. При ювенильном липофусцинозе, вызванном CLN3, электронная микроскопия тканей выявляет отложения, называемые профилями отпечатков пальцев, а обычная световая микроскопия мазка периферической крови может показать вакуоли в лимфоцитах.