При первичной недостаточности надпочечников врожденные или приобретенные поражения коры надпочечников препятствуют выработке кортизола, а часто и альдостерона (табл. 1). Приобретенная недостаточность надпочечников называется болезнью Аддисона. Дисфункция передней доли гипофиза или гипоталамуса может стать причиной дефицита кортикотропина (АКТГ) и привести к гипофункции коры надпочечников, что обозначается как вторичная недостаточность надпочечников; понятие третичной недостаточности надпочечников иногда используется для обозначения случаев, развившихся из-за дисфункции гипоталамуса (табл. 2).
Первичная недостаточность надпочечников у детей наиболее часто является результатом генетических заболеваний, которые часто, но не всегда, манифестируют в младенчестве, и реже — приобретенных проблем, напр. аутоиммунных заболеваний (табл. 3). Предрасположенность к аутоиммунным заболеваниям часто имеет генетическую основу, поэтому такое разделение не является абсолютным.
а) Наследственная этиология:
1. Врожденные нарушения синтеза стероидов. Наиболее частыми причинами недостаточности коры надпочечников в младенческом возрасте являются сольтеряющие формы врожденной гиперплазии надпочечников*. У 75% младенцев с недостаточностью 21-гидроксилазы, практически у всех младенцев с липоидной гиперплазией надпочечников и у большинства младенцев с недостаточностью 3β-гидроксистероид-дегидрогеназы в период новорожденности проявляются симптомы потери соли, потому что у них отсутствует способность синтезировать как кортизол, так и альдостерон.
P.S. В российской литературе — врожденная дисфункция коры надпочечников.
2. Врожденная гипоплазия надпочечников. Врожденная гипоплазия надпочечников (ВГН) является относительно частой причиной недостаточности надпочечников у мальчиков, наряду с врожденной гиперплазией надпочечников, аутоиммунными заболеваниями и адренолейкодистрофией (АЛД). Несмотря на название расстройства, ВГН — это преимущественно недостаточность развития дефинитивной зоны коры надпочечников; фетальная зона может быть относительно нормальной.
Следовательно, недостаточность надпочечников в целом становится очевидной по мере инволюции фетальной зоны, которая происходит после родов, начинаясь во младенчестве или в первые 2 года жизни, но иногда в более позднем детстве или даже во взрослом возрасте. В некоторых случаях дефицит альдостерона становится очевидным раньше, чем дефицит кортизола.
Причиной заболевания является мутация гена DAX1 (NR0B1) — члена семейства ядерных гормональных рецепторов, расположенного на Хр21. У мальчиков с ВГН часто не наступает пубертат вследствие гипогонадотропного гипогонадизма, вызванного аналогичным мутированным геном DAX1. Крипторхизм, иногда обнаруживаемый у этих мальчиков, вероятно, является ранним проявлением гипогонадотропного гипогонадизма, однако функция яичек у младенцев часто в пределах нормы, с типичным или даже необычно продолжительным выбросом тестостерона в течение 1-го месяца жизни.
ВГН иногда возникает как часть синдрома делеции генных последовательностей вместе с мышечной дистрофией Дюшенна, дефицитом глицерокиназы, когнитивными нарушениями или комбинацией этих состояний.
3. Другие генетические причины гипоплазии надпочечников. Фактор транскрипции SF-1 необходим для развития надпочечников и половых желез. У мальчиков с гетерозиготной мутацией в SF-1 (NR5A1) обнаруживается нарушение развития яичек, несмотря на наличие нормальной копии гена на др. хромосоме, они могут казаться девочками, подобно пациентам с липоидной гиперплазией надпочечников. Редко у таких пациентов также имеется недостаточность надпочечников.
Гипоплазия надпочечников также иногда наблюдается у пациентов с синдромом Паллистера-Холла, вызванным мутациями в онкогене GLI3.
4. Адренолейкодистрофия. При АЛД недостаточность коры надпочечников сочетается с демиелинизацией в ЦНС. В тканях и жидких средах организма обнаруживают высокие уровни очень длинноцепочечнных жирных кислот, что является результатом нарушения их β-окисления в пероксисомах.
Наиболее распространенной формой АЛД является Х-сцепленное заболевание с различными проявлениями. Наиболее часто клиническая картина представлена дегенеративным неврологическим заболеванием, проявляющимся в детстве или подростковом возрасте и прогрессирующим до тяжелой деменции и нарушения зрения, слуха, речи и походки, заканчивающимся смертью в течение нескольких лет. Неврологические симптомы могут быть едва заметными в начале, иногда состоящими только из поведенческих изменений или ухудшения успеваемости.
Генерализованная, но неполная алопеция, напоминающая таковую в результате XT, является характерным, но непостоянным признаком. Более легкой формой Х-сцепленной АЛД является адреномиелонейропатия, которая начинается в более позднем подростковом возрасте или в старшем юношеском возрасте. У пациентов м.б. признаки недостаточности надпочечников до, во время или после возникновения неврологических симптомов, часто их появление разделяют годы. Х-сцепленная АЛД является результатом мутаций в гене ABCD1, расположенном на Xq28. Этот ген кодирует трансмембранный транспортер, участвующий в доставке очень длинноцепочечных жирных кислот внутрь пероксисом.
У пациентов с Х-сцепленной АЛД было описано более 400 мутаций. Клинические фенотипы могут варьировать среди членов одной семьи, возможно, из-за генов-модификаторов или др. неизвестных факторов.
Корреляции между степенью неврологических нарушений и тяжестью недостаточности надпочечников нет. Доступны пренатальная диагностика при помощи анализа ДНК и семейный скрининг с использованием исследований очень длинноцепочечных жирных кислот и мутационного анализа. У женщин, которые являются гетерозиготными носительницами гена Х-сцепленной АЛД, могут развиваться симптомы в среднем возрасте или позднее; недостаточность надпочечников встречается редко.
АЛД новорожденных является редким АуР-заболеванием. У младенцев отмечается ухудшение неврологического состояния, также они имеют или приобретают признаки дисфункции коры надпочечников. Большинство пациентов имеют тяжелые, прогрессирующие когнитивные нарушения и умирают в возрасте до 5 лет. Это расстройство является разновидностью синдрома Цельвегера (цереброгепаторенального), при котором пероксисомы вообще не развиваются из-за мутаций в любом из нескольких генов (РЕХ5, РЕХ1, РЕХ10, РЕХ13 и РЕХ26), контролирующих развитие этой органеллы.
5. Семейная форма дефицита глюкокортикоидов. Семейная форма дефицита ГКС — форма хронической недостаточности надпочечников, характеризующаяся изолированным дефицитом ГКС, повышением уровня АКТГ и обычно нормальной продукцией альдостерона, хотя имеющиеся при большинстве других форм недостаточности надпочечников проявления потери соли иногда происходят. У пациентов в основном отмечается гипогликемия, судороги и усиление пигментации в течение первого десятилетия жизни. Заболевание поражает представителей обоих полов и наследуется по АуР-типу.
Отмечается выраженная атрофия коры надпочечников с относительно нетронутой клубочковой зоной. Примерно у 25% этих пациентов были описаны мутации в гене рецептора АКТГ (MCR2), большинство из которых влияют на транспорт молекул рецептора от эндоплазматической сети к поверхности клетки. Еще 20% случаев вызваны мутациями гена MRAP, который кодирует акцессорный белок рецептора меланокортина, требующийся для такой направленной миграции. Были идентифицированы мутации в новых генетических локусах, включая гомолог с недостаточностью комплекса поддержания минихромосомы 4 (МСМ4) и никотинамиднуклеотидтрансгидрогеназу (NNT).
Эти гены участвуют в репликации ДНК и антиоксидантной защите соответственно. У пациентов с мутациями МСМ4 также имеется отставание в росте, увеличение частоты поломок хромосом и дефицит естественных клеток-киллеров.
Другой синдром резистентности к АКТГ возникает в сочетании с ахалазией кардиального отдела желудка и алакримией (синдром трех «А», или синдром Оллгрова). У таких пациентов часто обнаруживается прогрессирующее неврологическое расстройство, которое включает вегетативную дисфункцию, умственную отсталость, моторную нейропатию и периодическую глухоту. Этот синдром также наследуется по АуР-типу, а ген AAAS картирован на хромосоме 12q13. Кодируемый белок, аладин, может помочь в регуляции ядерно-цитоплазматического транспорта др. белков.
6. Аутоиммунный полиэндокринный синдром типа I. Хотя аутоиммунная болезнь Аддисона наиболее часто возникает спорадически, она может развиться как часть двух синдромов, каждый из которых состоит из совокупности аутоиммунных расстройств. Аутоиммунный полиэндокринный синдром типа I (АПС-1), также известный как синдром аутоиммунной полиэндокринопатии-кандидоза-эктодермальной дистрофии (APECED), наследуется по менделевскому АуР-типу, тогда как АПС-2 (см. раздел «Аутоиммунная болезнь Аддисона» в этой главе) имеет сложное наследование.
Хронический кандидоз кожи и слизистых оболочек обычно является первым проявлением АПС-1, с последующим развитием гипопаратиреоза и затем болезни Аддисона, которая обычно возникает в раннем подростковом возрасте. Другие тесно связанные аутоиммунные расстройства включают недостаточность половых желез, алопецию, витилиго, кератопатию, гипоплазию зубной эмали, дистрофию ногтей, нарушение процессов всасывания в тонком кишечнике и хронический активный гепатит. Гипотиреоз и СД-1 возникают менее чем у 10% пациентов с данным нарушением.
Некоторые компоненты синдрома продолжают развиваться даже в течение пятого десятилетия жизни. У пациентов с АПС-1 могут обнаруживаться АТл к ферментам надпочечников семейства цитохрома Р450 — CYP21, CYP17 и CYP11A1. Наличие таких АТл свидетельствует о высокой вероятности развития болезни Аддисона или, у пациенток женского пола, о недостаточности яичников. Недостаточность надпочечников может быстро развиться при АПС-1; сообщалось о смерти пациентов с ранее поставленным диагнозом и необъяснимых смертях у сиблингов пациентов с АПС-1, что указывает на необходимость тщательного наблюдения за пациентами с АПС-1 (или любым ребенком с гипопаратиреозом неизвестной этиологии) и тщательной оценки сиблингов пациентов с этим расстройством, которые, предположительно, им не страдают.
Ген, поврежденный при АПС-1, обозначается как аутоиммунный регулятор-1 (AIRE1); он картирован на хромосоме 21q22.3. Ген AIRE1 кодирует фактор транскрипции, который контролирует экспрессию многих белков в пределах тимуса, тем самым играя критическую роль в формировании иммунологической толерантности. У пациентов с АПС-1 было описано множество различных мутаций в гене AIRE1, две из которых (делеция R257X и 3-bp) являются наиболее частыми. У одного родственника была обнаружена аутосомно-доминантная передача вследствие специфической миссенс-мутации (G228W).
7. Нарушения синтеза и метаболизма холестерина. У пациентов с нарушениями синтеза или метаболизма холестерина, включая абеталипопротеинемию с недостатком липопротеинов, содержащих липопротеины В (напр., липопротеины низкой плотности), и гомозиготную семейную гиперхолестеринемию с повреждением или отсутствием рецепторов липопротеинов низкой плотности, отмечается умеренное нарушение функции коры надпочечников. У пациентов с гетерозиготной семейной гиперхолестеринемией имеется нормальная функция коры надпочечников, которая не изменяется при лечении статинами (ингибитор ГМГ-КоА редуктазы).
У пациентов с синдромом Смита-Лемли-Опица — АуР-расстройством, проявляющимся аномалиями лица, микроцефалией, аномалиями конечностей и задержкой развития — была зарегистрирована недостаточность надпочечников. При синдроме Смита-Лемли-Опица были выявлены мутации в гене, кодирующем стерол-Δ7-редуктазу, картированном на хромосоме 11q12-q13, приводящие к нарушению конечной стадии синтеза холестерина с выраженным подъемом уровня 7-дегидрохолестерина, патологически низким уровнем холестерина и недостаточностью надпочечников. Болезнь Вольмана — редкое АуР-заболевание, вызванное мутациями в гене, кодирующем кислую липазу лизосом человека на хромосоме 10q23.2-23.3.
Эфиры холестерина накапливаются в лизосомах в большинстве систем органов, что приводит к органной недостаточности. У младенцев в течение 1-го или 2-го мес жизни отмечается гепатоспленомегалия, стеаторея, вздутие живота и снижение прибавки в весе и задержка роста. Имеются недостаточность и двусторонний кальциноз надпочечников, а смерть обычно наступает на первом году жизни.
8. Дефицит кортикостероид-связывающего глобулина и снижение кортизол-связывающей способности. Результатом дефицита кортикостероид-связывающего глобулина и снижения кортизол-связывающей способности является низкий уровень кортизола в плазме крови, при этом уровень свободного кортизола мочи и уровень АКТГ плазмы крови остаются нормальными. Сообщалось о высокой частоте развития гипотензии и утомляемости у некоторых взрослых с дефицитом кортикостероид-связывающего глобулина.
б) Приобретенные причины:
1. Аутоиммунная болезнь Аддисона. Наиболее частая причина болезни Аддисона — аутоиммунная деструкция желез. Железы могут быть настолько малы, что они не видны при аутопсии, причем в срезах при микроскопии обнаруживаются только остатки ткани. Обычно мозговое в-во не разрушено, а в области бывшей коры отмечается выраженная лимфоцитарная инфильтрация. При далеко зашедшем заболевании утрачены все функции коры надпочечников, но на ранних стадиях клинического течения может возникнуть дефицит кортизола.
У большинства пациентов обнаруживаются цитоплазматические АТл к АГн надпочечников в плазме крови; 21-гидроксилаза (CYP21) является наиболее часто встречающимся биохимически определяемым аутоАГн.
Болезнь Аддисона может возникать как компонент двух аутоиммунных полиэндокринных синдромов. Тип I (АПС-1) обсуждался выше. Аутоиммуный полиэндокринный синдром типа II (АПС-2) включает болезнь Аддисона с аутоиммунным заболеванием ЩЖ (синдром Шмидта) или СД-1 (синдром Карпентера). Могут развиваться недостаточность половых желез, витилиго, алопеция и хронический атрофический гастрит с пернициозной анемией или без нее. Частоты аллелей человеческого лейкоцитарного АГн (HLA)-DR3 и HLA-DR4 повышены у этих пациентов, что, по-видимому, повышает риск развития этого заболевания; определенные аллели в генах главного комплекса гистосовместимости класса I, связанных с цепью А и В (MICA и MICB), также ассоциированы с данным расстройством.
Связь полиморфизма генов, вовлеченных в другие аутоиммунные нарушения, с первичной недостаточностью надпочечников была нестабильна, при этом их вклад в ее патогенез следует рассматривать как неопределенный. К ним относятся главный комплекс гистосовместимости класса II, трансактиватор (СПТА), член А семейства 16 лектиновых доменов С-типа (CLEC16A) и нерецепторный белок тирозинфосфатаза тип 22 (PTPN22). Заболевание распространено среди женщин среднего возраста и может возникать во многих поколениях одной семьи. У этих пациентов также обнаруживаются АТл к АГн надпочечников, в частности АТл к ферментам CYP21, CYP17 и CYP11A1. Аутоиммунная недостаточность надпочечников может также наблюдаться у пациентов с целиакией и мутациями митохондриальных генов.
2. Инфекция. В прошлом туберкулез служил частой причиной деструкции надпочечников, однако в настоящее время он менее распространен. Наиболее частой причиной недостаточности надпочечников инфекционной природы является менингококцемия; адреналовый криз вследствие этой причины обозначают как синдром Уотерхауса-Фридериксена. У пациентов с ВИЧ-инфекцией могут наблюдаться разнообразные субклинические нарушения в гипоталамо-гипофизарно-надпочечниковой (ГГН) оси, однако явная недостаточность надпочечников встречается редко. Тем не менее ЛП, которые применяются в лечении ВИЧ-инфекции, могут влиять на гомеостаз гормонов надпочечников.
3. Лекарственные препараты. Кетоконазол — противогрибковый ЛП, ингибируя адреналовые ферменты, может вызывать недостаточность надпочечников. Митотан (o,p'-DDD), применяемый в лечении адренокортикальной карциномы и рефрактерного синдрома Кушинга, является цитотоксичным по отношению к коре надпочечников и также может вызывать изменения вненадпочечникового метаболизма кортизола. Признаки недостаточности надпочечников встречаются у значительного процента пациентов, которым проводилось лечение митотаном. Этомидат, применяемый для индукции и поддержания общего наркоза, ингибирует 11β-гидроксилазу (CYP11B1), а единственная индукционная доза может заблокировать синтез кортизола на 4-8 ч и более.
Это может вызвать сложности у пациентов, находящихся в глубоком стрессовом состоянии, особенно в случае повторного применения в условиях экстренной помощи. Абиратерона ацетат — ингибитор биосинтеза андрогенов, который применяется в лечении метастазирующей карциномы простаты, угнетает биосинтез кортизола, но не нарушает биосинтез кортикостерона. Этот ЛП в настоящее время не встречается в педиатрической практике. Рифампицин и противосудорожные ЛП, такие как фенитоин и фенобарбитал, хотя сами и не являются причиной недостаточности надпочечников, но снижают эффективность и биодоступность кортикостероидной заместительной терапии, индуцируя ферменты печени, принимающие участие в метаболизме стероидов.
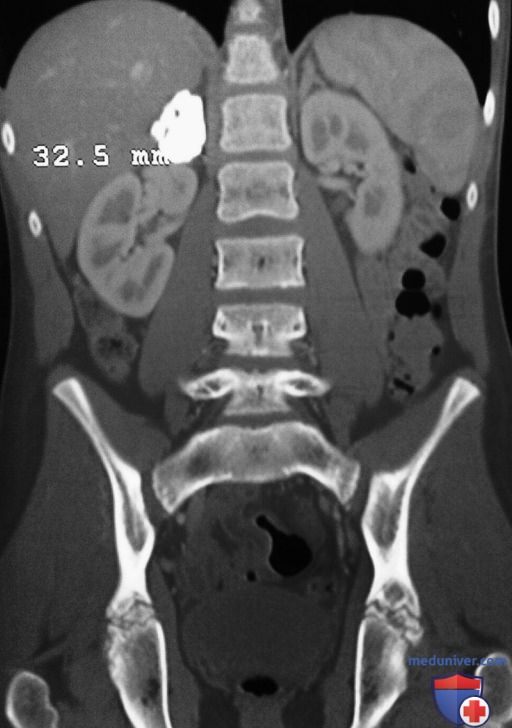
4. Кровоизлияние в надпочечники. Кровоизлияние в надпочечники может возникнуть в неонатальном периоде как последствие тяжелых родов (в особенности в тазовом предлежании), или его этиология м.б. неочевидна (рис. 1). Предполагаемая заболеваемость составляет 3:100 000 живорожденных. Кровоизлияние может быть достаточно обширным, чтобы привести к смерти от потери крови или гипоадренализма. Объемное образование в БП, анемия, необъяснимая желтуха или гематома мошонки могут быть ведущими симптомами. Часто кровоизлияние вначале протекает бессимптомно и выявляется позднее по наличию кальцинатов в надпочечниках. Также сообщалось о кровоизлиянии в надпочечники у плода.
Рисунок 1. Контрастно-усиленная компьютерная томограмма во фронтальной проекции, подтверждающая в/надпочечниковую локализацию округлого гиперденсного очага, что согласуется с крупным кальцинатом
В постнатальном периоде кровоизлияние в надпочечники наиболее часто возникает у пациентов, в отношении которых проводилось лечение антикоагулянтами. Оно также может быть результатом насилия над ребенком.
5. Клинические проявления. Первичная недостаточность надпочечников приводит к дефициту кортизола, а часто — и к дефициту альдостерона. Признаки и симптомы недостаточности надпочечников наиболее просто понять в контексте действия этих гормонов в норме (табл. 4).
Дефицит кортизола снижает сердечный выброс и тонус сосудов; более того, в отсутствие кортизола инотропные и прессорные эффекты катехоламинов, напр. адреналина, снижаются. Эти нарушения изначально проявляются ортостатической гипотензией у детей старшего возраста и могут прогрессировать до выраженного шока у пациентов любого возраста. Дефицит альдостерона, который приводит к гиповолемии вследствие снижения реабсорбции натрия в дистальных отделах нефрона, вызывает их обострение.
Гипотензия и снижение сердечного выброса снижают клубочковую фильтрацию и тем самым уменьшают способность почек выводить свободную воду. Вазопрессин (АДГ) секретируется задней долей гипофиза в ответ на гипотензию, а также как прямое следствие отсутствия подавления кортизолом. Эти факторы снижают осмоляльность плазмы крови и приводят, в частности, к гипонатриемии. Гипонатриемия также возникает в результате дефицита альдостерона и может быть намного тяжелее при одновременном дефиците кортизола и альдостерона.
Кроме гиповолемии и гипонатриемии дефицит альдостерона приводит к гиперкалиемии вследствие снижения экскреции калия в дистальных отделах нефрона. Дефицит только кортизола не приводит к гиперкалиемии.
Дефицит кортизола подавляет отрицательную обратную связь гипоталамуса и гипофиза, что приводит к увеличению секреции АКТГ. Гиперпигментация вызывается АКТГ и др. пептидными гормонами (γ-меланоцитстимулирующим гормоном), возникающими из предшественника АКТГ — проопиомеланокортина. У пациентов со светлым цветом лица кожа может приобретать бронзовый оттенок. Пигментация может быть более заметной в складках кожи, на слизистой оболочке и шрамах. У пациентов с темным цветом кожи ее проще всего оценивать на слизистой оболочке десен и щек.
Гипогликемия — характерный признак недостаточности надпочечников. Она часто сопровождается кетозом по мере того, как организм начинает использовать жирные кислоты в качестве альтернативного источника энергии. Кетоз усугубляется часто возникающими анорексией, тошнотой и рвотой.
Клиническая картина недостаточности надпочечников зависит от возраста больного, наличия нарушений секреции кортизола и альдостерона, а также в некоторой степени от этиологии, лежащей в ее основе. Наиболее частыми причинами в раннем младенческом возрасте являются врожденные нарушения биосинтеза стероидов, сепсис, ВГН и кровоизлияние в надпочечники. У младенцев отмечается относительно более высокая потребность в альдостероне, чем у детей более старшего возраста, что, вероятно, связано с незрелостью почек и низким содержанием натрия в человеческом грудном молоке и смесях для кормления. Гиперкалиемия, гипонатриемия и гипогликемия — ведущие признаки недостаточности надпочечников у младенцев. Кетоз имеется не всегда, т.к. кетоновые тела у младенцев образуются несколько хуже, чем у детей более старшего возраста.
Гиперпигментация обычно не наблюдается, потому что ее развитие занимает недели или месяцы; также очевидно, что у младенцев трудно распознать ортостатическую гипотензию.
Младенцы могут заболеть очень быстро. До развития критических электролитных нарушений может пройти всего несколько дней с момента появления снижения активности, отсутствия аппетита или рвоты.
У детей более старшего возраста с болезнью Аддисона симптомы включают мышечную слабость, чувство общего недомогания, отсутствие аппетита, рвоту, похудение и ортостатическую гипотензию. Они могут иметь скрытое начало. Нередко можно ретроспективно по данным анамнеза выявить эпизодическое возникновение этих симптомов в течение многих лет только во время интеркуррентных заболеваний. У таких пациентов может возникнуть острая декомпенсация (адреналовый криз) во время относительно нетяжелого инфекционного заболевания. Некоторым из этих пациентов изначально был установлен неверный диагноз синдрома хронической усталости, синдрома перенесенного мононуклеоза, хронической болезни Лайма или психического заболевания (депрессии или нервной анорексии).
Гиперпигментация наблюдается часто, но не всегда. Гипонатриемия обнаруживается на момент диагностики почти у 90% пациентов. Гиперкалиемия обычно возникает на более поздних этапах течения заболевания у детей более старшего возраста, чем младенческий, и отмечается только у половины пациентов на момент диагностики. Нормальный уровень калия никогда не должен исключать первичную недостаточность надпочечников.
Часто имеются гипогликемия и кетоз. Т.о., по клинической картине данное заболевание легко принять за гастроэнтерит или др. острое инфекционное заболевание. Хронический характер симптомов может насторожить клинициста в плане возможного наличия болезни Аддисона, но этот диагноз следует рассмотреть у всех детей с ортостатической гипотензией, гипонатриемией, гипогликемией и кетозом. При первичной недостаточности надпочечников с дефицитом минералокортикоидов наблюдается пристрастие к соли. При дефиците ГКС могут отмечаться усталость, миалгии, лихорадка, эозинофилия, лимфоцитоз, гиперкальциемия и анемия.
в) Лабораторные признаки. Гипогликемия, кетоз, гипонатриемия и гиперкалиемия уже обсуждались. Для быстрого выявления гиперкалиемии у ребенка в критическом состоянии пригодится ЭКГ. Обычно отмечается ацидоз, а уровень АМК повышен, если пациент находится в состоянии обезвоживания.
Уровни кортизола иногда находятся в нижней части нормального диапазона, но всегда низкие, когда решается вопрос о степени тяжести болезни пациента. При первичной недостаточности надпочечников отмечается высокий уровень АКТГ, однако его определение в лабораторных условиях может занять некоторое время. Сходным образом уровень альдостерона может быть в пределах нормального диапазона, но если учитывать наличие у пациента гипонатриемии, гиперкалиемии и гиповолемии, его уровень слишком низок. Активность ренина плазмы крови повышена. Количество эозинофилов в крови может быть повышено, хотя в диагностическом плане это редко имеет значение.
Экскреция натрия и хлора с мочой повышена, содержание калия в моче снижено, но это сложно оценить в случайных образцах мочи. Точная интерпретация содержания электролитов в моче требует более продолжительного (суточного) сбора мочи и знания потребления натрия и калия пациентом.
Наиболее точным исследованием для выявления недостаточности надпочечников является измерение уровня кортизола в сыворотке крови до и после введения АКТГ; его содержание в покое низкое и после введения АКТГ не увеличивается, как в норме (табл. 5). Иногда нормальные уровни в покое, которые не повышаются после введения АКТГ, свидетельствуют об отсутствии адренокортикального резерва. Низкий начальный уровень с последующим выраженным ответом на введение АКТГ может говорить о вторичной недостаточности надпочечников. Традиционно этот анализ проводился путем измерения уровней кортизола до и спустя 30 или 60 мин после введения 0,250 мг козинтропина (АКТГ 1-24) в виде быстрой в/в-инфузии. Уровень альдостерона транзиторно повышается в ответ на введение такой дозы АКТГ и также может быть измерен.
Проба с низкой дозой (1 мкг АКТГ 1-24/1,73 м2) является более чувствительным анализом для определения гипофизарно-надпочечникового резерва, но в некоторой степени менее специфична (больше ложноположительных результатов).
Примечание. В РФ при отсутствии препаратов АКТГ короткого действия предлагается использовать депо-форму АКТГ — тетракозактид в/м 1 мг с последующим исследованием уровней кортизола через 10-12 и 24 ч после инъекции.
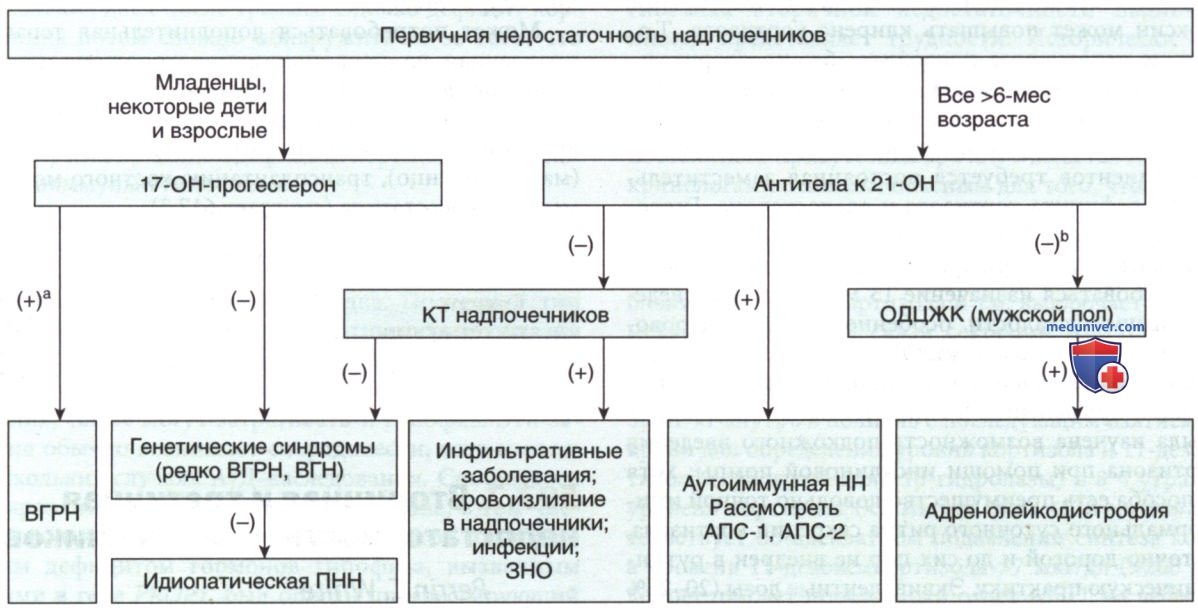
г) Дифференциальная диагностика. По клинической картине болезнь Аддисона часто необходимо дифференцировать с более острыми состояниями, такими как гастроэнтерит с обезвоживанием или сепсис. Дополнительное исследование направлено на выявление специфических причин недостаточности надпочечников. При подозрении на врожденную гиперплазию надпочечников необходимо измерить уровни предшественников кортизола (17-гидроксипрогестерон) в сыворотке крови наряду с измерением уровня кортизола в ходе теста со стимуляцией АКТГ (рис. 2).
Рисунок 2. Алгоритм диагностического подхода к пациенту с первичной недостаточностью надпочечников. Наиболее частые причины первичной недостаточности надпочечников — аутоиммунная деструкция коры надпочечников у взрослых и врожденная гиперплазия надпочечников у детей. Скрининг для выявлениях этих причин м.б. проведен с использованием определения антител к 21-гидроксилазе и базального уровня 17-гидроксипрогестерона в сыворотке крови соответственно. Мальчикам с отрицательным результатом анализа на антитела к 21-гидроксилазе необходимо провести исследование на наличие адренолейкодистрофии с определением ОДЦЖК в плазме крови. Если эти диагнозы исключены, то компьютерная томография надпочечников может выявить признаки инфильтративных процессов или метастазов в надпочечниках, но у детей и подростков такое встречается редко. Клиническая картина и семейный анамнез пациента могут сократить некоторые этапы диагностического алгоритма или помочь предположить наличие специфических генетических синдромов. Последние включают подтипы аутоиммунных полигландулярных синдромов или специфические редкие генетические нарушения, когда недостаточность надпочечников является частью более широкого фенотипа
Диагностическим критерием АЛД является повышение уровня очень длинноцепочечных жирных кислот. При помощи прямого генетического анализа могут быть выявлены многие генетические причины первичной недостаточности надпочечников, но получение его результатов может занять много недель. Наличие антител к антигенам надпочечников позволяет предположить аутоиммунный патогенез. Пациентов с аутоиммунной болезнью Аддисона необходимо тщательно наблюдать на предмет развития других аутоиммунных заболеваний. У детей она наиболее часто сочетается с гипопаратиреозом, который следует заподозрить при наличии гипокальциемии и повышения уровня фосфатов.
УЗИ (должен проводить опытный врач), КТ или МРТ могут помочь в определении размера надпочечников.
д) Лечение. Лечение острой недостаточности надпочечников должно быть немедленным и интенсивным. Если диагноз недостаточности надпочечников не был установлен, необходимо получить образец крови до начала лечения для определения уровня электролитов, глюкозы, АКТГ, кортизола, альдостерона и активности ренина плазмы. Если состояние пациента позволяет, пока проводится начальная агрессивная инфузионная терапия, м.б. проведена проба со стимуляцией АКТГ. Для коррекции гипогликемии, гиповолемии и гипонатриемии в/в вводят раствор декстрозы («Глюкозы») 5% в 0,9% растворе натрия хлорида. Следует избегать применения гипотонических растворов [напр., 5% раствора декстрозы («Глюкозы») в воде или 0,2% растворе натрия хлорида], т.к. они могут спровоцировать или усугубить гипонатриемию.
При выраженной гиперкалиемии может потребоваться лечение с в/в-введением кальция и/или бикарбонатов, применением интраректальной калий-связывающей смолы (полистиролсульфонат натрия, каексалат) или в/в-введением декстрозы («Глюкозы») и инсулина. Водорастворимые формы гидрокортизона, напр. гидрокортизон сукцинат натрия, необходимо вводить в/в. Не более 10 мг для младенцев, 25 мг для детей младшего возраста, 50 мг для детей старшего возраста и 100 мг для подростков следует вводить в виде болюса (В РФ 100 мг/м2) и аналогичное общее количество вводить разделенными дозами с интервалом в 6 ч в течение первых 24 ч (в РФ 100-200 мг/м2/сут в/в-капельно 1-2). Эти дозы могут быть уменьшены в течение следующих суток при удовлетворительном прогрессе.
Адекватное насыщение жидкостью и натрием достигается при помощи в/в-введения раствора натрия хлорида, чему способствует минералокортикоидный эффект высоких доз гидрокортизона.
Особое внимание необходимо уделить пациенту с сочетанием недостаточности надпочечников и гипотиреоза, т.к. тироксин может повышать клиренс кортизола. Т.о., если лечение гипотиреоза проводится без предварительного назначения адекватной заместительной терапии ГКС, можно спровоцировать адреналовый криз.
После того как острые проявления устранены, большинству пациентов требуется постоянная заместительная терапия дефицита кортизола и альдостерона. Гидрокортизон (кортизол) м.б. назначен внутрь 10 мг/м2/сут (в РФ 8-10 мг/м2/сут) в три приема; некоторым пациентам может потребоваться назначение 15 мг/м2/сут для сведения к минимуму усталости, особенно утренней. Проводятся клинические исследования ЛП гидрокортизона с отсроченным высвобождением, но они пока не доступны для широкого применения. В клинических исследованиях также была изучена возможность подкожного введения гидрокортизона при помощи инсулиновой помпы; хотя у этого способа есть преимущество довольно точной имитации нормального суточного ритма секреции кортизола, он достаточно дорогой и до сих пор не внедрен в рутинную клиническую практику.
Эквивалентные дозы (20-25% от дозы гидрокортизона) преднизона или преднизолона м.б. разделены на два приема. Для контроля достаточности заместительной терапии ГКС при первичной недостаточности надпочечников м.б. использовано измерение уровня АКТГ (в РФ исследование уровня АКТГ для контроля дозы ГКС не предполагается); при врожденной гиперплазии надпочечников вместо него используют определение уровня гормонов-предшественников. Образцы крови для контроля должны быть получены в одно и то же время дня и в соответствии с приемом гидрокортизона (т.е. до или после приема). Нормализация уровня АКТГ не нужна и может потребовать применения чрезмерных доз гидрокортизона; в целом, удовлетворительными считаются уровни АКТГ в утренние часы на верхней границе нормального диапазона до значений, в 3-4 раза превышающих нормальные.
Поскольку у пациентов, которые не получали лечение совсем или значительно недополучали его, во время относительно нетяжелой болезни может возникнуть острая декомпенсация, то оценка симптомов (или отсутствие таковых) не должна использоваться как замена контроля по биохимическим показателям. При стрессовых ситуациях, напр. во время инфекционных заболеваний или малых хирургических вмешательств, доза гидрокортизона должна быть увеличена в 2-3 раза.
Крупные операции под общим наркозом требуют в/в-введения высоких доз гидрокортизона, аналогичных таковым, которые применяются при острой недостаточности надпочечников.
При наличии дефицита альдостерона применяется синтетический минералокортикоид флудрокортизон 0,05-0,2 мг/сут внутрь. Измерение активности ренина плазмы крови используется для контроля достаточности заместительной терапии минералокортикоидами. Хроническая передозировка ГКС ведет к ожирению, низкорослости и остеопорозу, в то время как результатом передозировки флудрокортизона является АГ и иногда гипокалиемия.
Вопрос о необходимости замещения дефицита дегидроэпиандростерона (ДГЭА) у взрослых остается спорным; у детей до периода полового созревания большое количество ДГЭА в норме не секретируется. Многие взрослые пациенты с болезнью Аддисона жалуются на состояние пониженной энергии, и замещение ДГЭА может решить эту проблему, особенно у жнщин, у которых андрогены надпочечников составляют -50% от общей секреции андрогенов.
Может потребоваться дополнительная терапия, направленная на первопричину недостаточности надпочечников, в отношении инфекций и некоторых метаболических нарушений. Подходы к лечению АЛД включают введение триолеата глицерина и триэруката глицерина (масло Лоренцо), трансплантацию костного мозга и применение ловастатина.