а) Пероксисомные нарушения. Нарушения, связанные с жирными кислотами с очень длинной цепью (ОДЦЖК), относятся к более широкой группе пероксисомных заболеваний. Пероксисомные заболевания считаются генетически обусловленными нарушениями. Они связаны с нарушением формирования или структуры пероксисом или с нарушением функции единственного белка, который в норме имеется в этой органелле. Эти нарушения приводят к тяжелой инвалидизации в детском возрасте. Они возникают намного чаще и представлены более широким диапазоном фенотипов, чем распознавали в прошлом. Многие, но не все пероксисомные нарушения сопровождаются повышением уровней ОДЦЖК. Мы обсудим более широкую группу пероксисомных нарушений, основываясь на их проявлениях в детском возрасте.
1. Этиология. Пероксисомные нарушения подразделяют на две большие категории (табл. 2). При нарушениях биогенеза пероксисом (НБП) основной дефект заключается в отсутствии переноса одного или нескольких белков в органеллу. Др. группа нарушений связана с единственным пероксисомным белком (одноферментные дефекты). Пероксисомы имеются во всех клетках, за исключением зрелых эритроцитов. Они относятся к субклеточным органеллам и окружены однослойной мембраной. Идентифицировано >50 ферментов пероксисом. Некоторые ферменты участвуют в синтезе и расщеплении пероксида водорода, др. — в метаболизме липидов и аминокислот.
Большинство пероксисомных ферментов сначала синтезируются в зрелой форме на свободных полирибосомах и выходят в цитоплазму. Белки, предназначенные для пероксисом, содержат специфические направляющие аминокислотные последовательности (peroxisome targeting sequences, PTS). Большинство пероксисомных матриксных белков содержат PTS1 — последовательность из трех аминокислот на карбоксильном конце. PTS2 представляет собой аминоконцевую последовательность. Она имеет критическое значение для импорта ферментов, участвующих в метаболизме плазмалогенов и жирных кислот с разветвленной цепью. Импорт белков включает сложные последовательности реакций, в которых участвует не менее 23 отдельных протеинов. Эти белки называются пероксинами, кодируются генами РЕХ.
2. Эпидемиология. Все пероксисомные нарушения, перечисленные в табл. 2, за исключением Х-сцепленной адренолейкодистрофии (АЛД), относятся к АуР-заболеваниям. АЛД является наиболее распространенным пероксисомным нарушением. Расчетный показатель заболеваемости составляет 1:17 000 живорожденных. Суммарный показатель заболеваемости для др. пероксисомных нарушений оценивается в 1:50 000 живорожденных. Однако ожидается, что при более широком охвате детей неонатальным скринингом реальные показатели заболеваемости для всех нарушений, связанных с жирными кислотами с очень длинной цепью, будут установлены более точно.
3. Патология. Для нарушения биогенеза пероксисом патогномоничным является отсутствие или сокращение числа пероксисом в клетках. При большинстве нарушений мембранные мешки содержат интегральные мембранные белки пероксисом. В этих белках отсутствует нормальный комплемент матриксных белков. Такие мешки называют «призраками» пероксисом. Патологические изменения наблюдаются в большинстве органов. Отмечаются выраженные характерные нарушения миграции нейронов, мелкоузловой цирроз печени, кисты почек, точечная хондродисплазия, нейросенсорная тугоухость, ретинопатия, врожденные заболевания сердца и признаки дисморфизма.
4. Патогенез. Все патологические изменения, вероятно, связаны с недостаточностью биогенеза пероксисом, сопровождающейся недостаточностью множества ферментов. При НБП множество пероксисомных ферментов не выполняют свои функции (табл. 3). Ферменты, уровень которых снижен или равен нулю, синтезируются, но слишком быстро распадаются, поскольку за пределами пероксисомы защита у них может отсутствовать. Каким образом нарушение функций пероксисом приводит к широко распространенным патологическим проявлениям, неясно.
При НБП выявлены мутации в 12 разных генах РЕХ. Характер и выраженность патологических признаков варьируют в зависимости от природы и степени нарушений импорта. Идентифицированы дефекты генов, приводящие к таким расстройствам, как синдром Цельвегера, неонатальная адренолейкодистрофия, инфантильная форма болезни Рефсума и РТХД. Эти расстройства получили свои названия до того, как была обнаружена их связь с пероксисомами. Считается, что первые три заболевания формируют клинический континуум. Синдром Цельвегера — самое тяжелое расстройство, инфантильная форма болезни Рефсума — наименее тяжелое, а неонатальная АЛД — промежуточное. Эти заболевания могут развиваться при мутациях в любом из 11 генов, участвующих в сборке пероксисом. По клиническим признакам невозможно различить, в каком конкретно гене имеется дефект.
Тяжесть клинических проявлений варьирует в зависимости от степени нарушения импорта белка. Мутации, полностью устраняющие импорт белка, часто сопровождаются фенотипом синдрома Цельвегера, тогда как миссенс-мутации, при которых функция импорта в некоторой степени сохраняется, приводят к несколько менее тяжелым фенотипическим проявлениям. Дефект в РЕХ7, отвечающем за импорт белков, которые утилизируют PTS2, ассоциируется с РТХД. Для дефектов РЕХ7, при которых импорт частично сохраняется, характерны более легкие фенотипические проявления, некоторые из них напоминают классическую (взрослую) форму болезни Рефсума.
Генетические расстройства с вовлечением одного из пероксисомных ферментов, как правило, имеют более ограниченные клинические проявления и связаны с одним биохим. дефектом. Первичная недостаточность надпочечников при АЛД обусловлена накоплением ОДЦЖК в коре надпочечников, а периферическая невропатия при взрослой форме болезни Рефсума — накоплением фитановой кислоты в шванновских клетках и миелине.
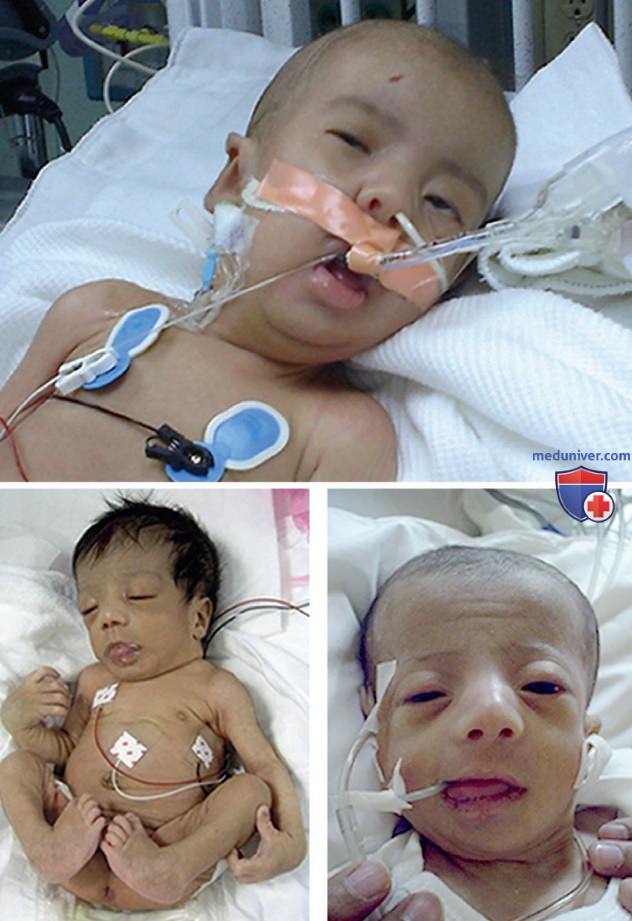
- Нарушение спектра синдрома Цельвегера. У новорожденных с синдромом Цельвегера имеются соответствующие бросающиеся в глаза отклонения. Центральное значение в диагностике имеют типичные черты лица (высокий лоб, наружный угол глаза выше внутреннего, гипоплазия надбровных дуг и эпикантус; рис. 3), выраженная слабость и гипотония, неонатальные судороги и офтальмологические нарушения. В связи с наличием гипотонии и характерных краниофациальных признаков можно заподозрить синдром Дауна. Младенцы с синдромом Цельвегера редко живут больше нескольких месяцев. Более чем у 90% наблюдают задержку роста в постнатальный период. В табл. 4 перечислены основные клинические отклонения.
Рисунок 3. Синдром Цельвегера. Три новорожденных с синдромом Цельвегера. Обратите внимание на гипотонию, высокий лоб со слабовыраженными надбровными дугами, вывернутые вперед ноздри и легкую микрогнатию, а также эквиноварусную деформацию стоп и контрактуру коленных суставов
У пациентов с неонатальной АЛД краниофациальные особенности выражены меньше. Неонатальные судороги возникают часто. Отмечаются некоторая задержка психомоторного развития, тяжелые нарушения интеллекта. После 3-5 лет развитие может регрессировать. Вероятно, это объясняется прогрессирующей лейкодистрофией. Неизменно всегда наблюдаются гепатомегалия, нарушение функции печени, пигментная дегенерация сетчатки и тяжелые нарушения слуха. Функция коры надпочечников, как правило, нарушена. Может потребоваться заместительная гормонотерапия. Точечная хондродисплазия и кисты почек отсутствуют.
Пациенты с инфантильной формой болезни Рефсума доживают до взрослого возраста. Они могут ходить, но походка м.б. неуверенной и неуклюжей. Когнитивные функции в большинстве случаев нарушены, однако возможности точной оценки, как правило, ограничены в связи с нарушениями зрения и слуха. Почти у всех пациентов отмечается некоторая степень нейросенсорной тугоухости и пигментной дегенерации сетчатки. У них имеются умеренно выраженные признаки дисморфизма, напр., эпикантус, плоская переносица и низкое расположение ушных раковин. Нередко рано возникают гипотония и гепатомегалия с нарушением функции печени. У многих пациентов в плазме умеренно снижены уровни ХС, ЛПВП и ЛПНП. Точечная хондродисплазия и кортикальные кисты почек отсутствуют.
Посмертное исследование при инфантильной форме болезни Рефсума выявило мелкоузловой цирроз печени и маленькие гипоплазированные надпочечники. В ГМ мальформаций не обнаружено, за исключением тяжелой гипоплазии зернистого слоя коры мозжечка и эктопического расположения клеток Пуркинье в молекулярном слое. Тип наследования АуР.
У некоторых пациентов с НБП наблюдаются менее выраженные и атипичные фенотипы. Они м.б. представлены периферической невропатией или ретинопатией, нарушениями зрения или катарактой у детей, подростков или взрослых. У таких пациентов диагностируют болезнь Шарко-Мари-Тута или синдром Ашера. Некоторые пациенты доживают до 50 лет. Дефекты РЕХ7, на фоне которых чаще всего развивается РТХД, могут также приводить к менее выраженному фенотипу, который по клиническим проявлениям схож со взрослой формой синдрома Рефсума.
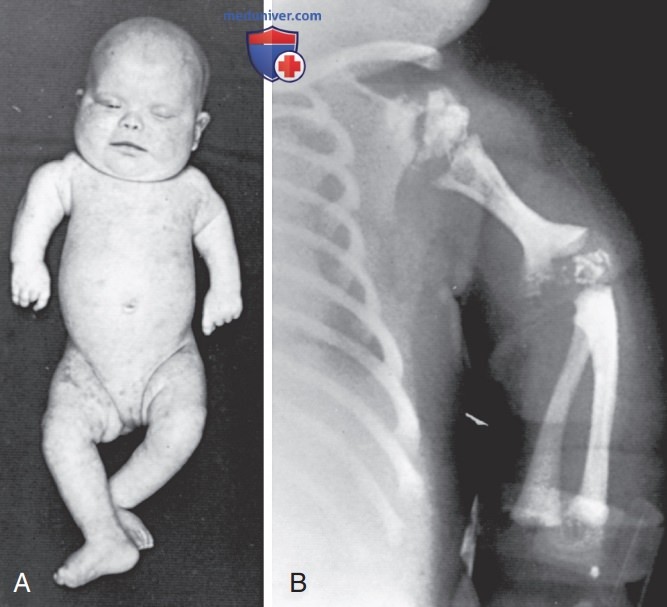
- Ризомелическая точечная хондродисплазия. Для РТХД характерны точечные фокусы кальцификации гиалинового хряща. Это заболевание сопровождается карликовостью, катарактами (72%) и множественными деформациями, связанными с контрактурами. В телах позвонков имеются фронтальные расщелины, заполненные хрящевой тканью. Они возникают в результате остановки эмбрионального развития. Проксимальные отделы конечностей непропорционально короткие (рис. 4). При рентгенографии обнаруживают укорочение костей проксимальных отделов конечностей, чашеобразную деформацию метафизов и нарушение оссификации (рис. 4). Рост, МТ и окружность головы — ниже 3-го процентиля. У таких детей отмечаются тяжелые нарушения интеллекта. У 25% пациентов имеются кожные изменения, подобные тем, которые наблюдают при ихтиозиформной эритродермии.
Рисунок 4. А — новорожденный с ризомелической точечной хондродисплазией. Обратите внимание на выраженное укорочение проксимальных отделов конечностей, низкое расположение переносицы, гипертелоризм и участки крупнопластинчатого шелушения на коже; В — обратите внимание на явное укорочение плечевой кости и точечную кальцификацию эпифизов плечевых и локтевых суставов
- Изолированные дефекты окисления жирных кислот в пероксисомах. Дефицит ацил-КоА-оксидазы и дефицит бифункционального фермента, входящие в группу одноферментных нарушений, охватывают один ферментный этап пероксисомного окисления жирных кислот. Дефекты бифункционального фермента встречаются часто. Их обнаруживают у 5% пациентов, у которых изначально подозревали заболевания спектра синдрома Зельвегера. У пациентов с изолированным дефицитом ацил-КоА-оксидазы отмечаются сходные, но несколько менее выраженные фенотипические проявления. Такие пациенты обращают на себя внимание в связи с развитием лейкодистрофии в раннем детском возрасте.
- Изолированные дефекты синтеза плазмалогенов. Плазмалогены — это липиды, в которых первый атом углерода в молекуле глицерина связан со спиртом, а не с жирной кислотой. Они синтезируются в результате сложной серии реакций. Первые два этапа синтеза катализируют пероксисомные ферменты дигидроксиацетонфосфаталкилтрансфераза и синтаза. Дефицит каждого из этих ферментов приводит к развитию фенотипа, который по клиническим проявлениям неотличим от нарушения пероксисомного импорта РТХД. Причиной последнего является дефект РЕХ7, рецептора PTS2. При РТХД, как и при одноферментных нарушениях, наблюдается выраженный дефицит плазмалогенов, кроме этого, имеются дефекты окисления фитановой кислоты. Т.о., указанные моногенные заболевания проявляются полным фенотипом РТХД, и это свидетельствует о том, что для формирования такого фенотипа достаточно дефицита плазмалогенов.
- Взрослая (классическая) форма болезни Рефсума. Дефектный фермент (фитаноил-КоА-гидроксилаза) располагается в пероксисомах. Болезнь Рефсума проявляется зрительными нарушениями, связанными с пигментным ретинитом, аносмией, ихтиозом, периферической невропатией, атаксией, в редких случаях — нарушениями ритма сердца. В отличие от инфантильной формы болезни Рефсума, когнитивные функции не страдают, врожденные мальформации также отсутствуют. Во многих случаях болезнь Рефсума не проявляется до молодого возраста, однако зрительные нарушения, напр. ночная слепота, ихтиоз и периферическая невропатия, могут присутствовать уже в детском и подростковом возрасте.
Большое значение имеет ранняя диагностика, поскольку раннее введение диеты с ограничением фитановой кислоты может обратить вспять периферическую невропатию и предотвратить прогрессирование проявлений со стороны зрительной и ЦНС. Фенотип взрослой формы болезни Рефсума может также развиваться при дефектах РЕХ7.
- Недостаточность 2-метилацил-КоА-рацемазы. Причиной этого расстройства является ферментный дефект, приводящий к накоплению жирных кислот с разветвленной цепью (фитановой и пристановой кислот), а также желчных кислот. Заболевание проявляется типичной периферической невропатией с началом клинической картины во взрослом возрасте. Кроме того, может развиваться пигментная дегенерация сетчатки.
5. Лабораторные признаки. Диагноз пероксисомного нарушения часто устанавливают по отклонениям биохимических показателей, а затем подтверждают результатами генетических исследований.
Отклонения биохим. показателей, характерные для пероксисомных нарушений, которые можно определить с помощью широкодоступных методов исследования, приведены в табл. 5. Чаще всего используют такой метод исследования, как определение уровней ОДЦЖК в плазме крови. Следует подчеркнуть, что уровни ОДЦЖК плазмы повышены у многих пациентов с пероксисомными нарушениями, но не у всех. Наиболее значимым исключением является РТХД, при которой уровни ОДЦЖК не отклоняются от нормы, однако определяются повышенные уровни фитановой кислоты плазмы и пониженные уровни плазмалогенов эритроцитов. При остальных пероксисомных нарушениях отклонения биохим. показателей выражены еще меньше. Исходя из этого, в рекомендованную панель тестов входит определение уровней ОДЦЖК, фитановой, пристановой и пипеколиновой кислот плазмы, а также уровней плазмалогенов эритроцитов.
Тандемная масспектрометрия также является подходящим методом количественного определения желчных кислот в плазме крови и моче. Эта панель исследований позволяет определить большинство пероксисомных нарушений, и для ее проведения требуется очень небольшое количество венозной крови. Кроме того, при нормальных результатах исследования наличие типичных пероксисомных нарушений очень маловероятно. Для установления клинического диагноза нередко достаточно результатов биохим. исследований и клинической картины. Разработаны методы с использованием сухих пятен крови. Эти методы включены в программу неонатального скрининга.
Следующим этапом является молекулярная ДНК-диагностика. Во многих клинических лабораториях имеется возможность определения пероксисомных панелей с использованием технологий нового поколения. В некоторых случаях диагноз устанавливают по результатам полноэкзомного секвенирования и по патогенетической природе повреждений, а затем подтверждают результатами биохим. исследований.
Определение молекулярного дефекта у пробанда имеет огромное значение для выявления патологии у бессимптомных носителей и ускоряет пренатальную диагностику. У пациентов с дефектами РЕХ1 описание мутаций может иметь прогностическое значение. Этот дефект имеется у 60% пациентов с НБП. Примерно в половине случаев при дефекте РЕХ1 имеется аллель G843D, что сопровождается значительно менее выраженными фенотипическими проявлениями, чем при др. мутациях.
6. Диагностика. Несколько неинвазивных методов лабораторного исследования позволяют точно и в ранние сроки установить диагноз пероксисомных нарушений (см. табл. 5). Трудность при НБП заключается в ДД этого заболевания с большим разнообразием др. состояний, вызывающих гипотонию, судороги, задержку роста или признаки дисморфизма. Опытные клиницисты быстро распознают классический синдром Цельвегера по клиническим проявлениям. Однако у пациентов с более легкими формами НБП часто наблюдается неполный спектр клинических проявлений заболевания. В таких случаях диагноз можно установить только по результатам лабораторных исследований.
Проведение диагностических исследований обосновано при наличии таких клинических признаков, как нарушения интеллекта, слабость и гипотония, признаки дисморфизма, неонатальные судороги, ретинопатия, глаукома или катаракта, снижение слуха, гепатомегалия и нарушение функции печени, точечная хондродисплазия.
Наличие одного или нескольких отклонений из перечисленных увеличивает вероятность указанного диагноза. Описаны также более легкие атипичные формы, которые проявляются периферической невропатией.
Некоторые пациенты с изолированными дефектами пероксисомного окисления жирных кислот напоминают пациентов с нарушениями спектра синдрома Цельвегера. Таких пациентов можно выявить по ненормально высоким уровням ОДЦЖК.
Пациентов с РТХД необходимо отличать от пациентов с др. причинами точечной хондродисплазии. Диагноз РТХД можно заподозрить по клиническим признакам — коротким конечностям, задержке развития, а также ихтиозу. При проведении лабораторных исследований наибольшее значение имеют патологически низкие уровни плазмалогенов в эритроцитах и изменение структуры РЕХ7.
7. Осложнения. У пациентов с синдромом Цельвегера имеется множество инвалидизирующих отклонений, в т.ч. нарушение мышечного тонуса и глотания, нарушения со стороны сердца, заболевания печени и судороги. Для лечения этих состояний применяют симптоматическую терапию, однако прогноз при них неблагоприятный и большинство пациентов умирают в первый год жизни. У пациентов с РТХД также имеется множество системных и неврологических проблем. Кроме того, у них может развиваться тетрапарез, обусловленный сдавлением основания ГМ.
8. Лечение. Наиболее эффективным методом терапии при взрослой форме болезни Рефсума является диета с ограничением фитановой кислоты. Однако этот метод действует только при указанной патологии.
У пациентов с несколько менее тяжелыми вариантами нарушений пероксисомного импорта эффективны вмешательства на ранних стадиях с использованием мульти-дисциплинарного подхода, в т.ч. лечебной физкультуры и трудотерапии, использование слуховых аппаратов или кохлеарных имплантов, вспомогательных и альтернативных способов коммуникации, диетотерапии, а также поддержки семьи. Хотя у большинства пациентов функциональные возможности так и не достигают нормальных значений, у некоторых из них наблюдается стабильное состояние в подростковом возрасте или в молодом возрасте старше двадцати лет.
Некоторые из вторичных отклонений биохимических показателей пытаются смягчить путем перорального применения докозагексаеновой кислоты (ДГА). У пациентов с нарушениями биогенеза пероксисом отмечают значительное снижение уровня ДГА, а такая терапия нормализует уровни ДГА плазмы. Имеются неподтвержденные сообщения о клиническом улучшении на фоне терапии ДГА, однако в плацебо-РКИ эффективность такой терапии не была подтверждена.
9. Генетическое консультирование. Все обсуждавшиеся пероксисомные нарушения можно диагностировать пренатально. Пренатальная диагностика с использованием ворсинок хориона или образцов, полученных путем амниоцентеза, как правило, основывается на генетическом исследовании в случаях, когда известно о наличии изменений, однако биохим. показатели можно определять с помощью тех же методов, которые описаны для постнатальной диагностики (см. табл. 5). Повторный риск рождения больного ребенка составляет 25%, поэтому пары, имеющие детей с такой патологией, следует информировать о возможностях пренатальной диагностики.
б) Адренолейкодистрофия (АЛД). АЛД — Х-сцепленное расстройство, сопровождающееся накоплением насыщенных ОДЦЖК и прогрессирующей дисфункцией коры надпочечников и НС. Это наиболее распространенное пероксисомное нарушение.
1. Этиология. Основное отклонение биохим. показателей при АЛД — накопление в тканях насыщенных ОДЦЖК с длинной цепью из 24 углеродных атомов и более. Наиболее выраженным и характерным изменением является избыток гексакозановой кислоты (С26:0). Накопление жирных кислот связано генетически обусловленным дефицитом деградации жирных кислот в пероксисомах. Дефектный ген (ABCD1) кодирует пероксисомный мембранный белок (ALDP, белок АЛД). Многие изменения в гене ABCD1 определены как патогенные. Приблизительно половина из них относятся к приватным мутациям или встречаются только у членов конкретной семьи. Имеется курируемая база данных.
По-видимому, механизм накопления ОДЦЖК при дефектах белка АЛД заключается в нарушении транспорта насыщенных жирных кислот в пероксисомы. В результате длина углеродных цепей жирных кислот продолжает прогрессивно нарастать.
2. Эпидемиология. Минимальный показатель заболеваемости АЛД у мужчин составляет 1:21 000. Суммарный показатель заболеваемости АЛД у мужчин и гетерозиготных женщин в общей популяции оценивается в 1:17 000. Заболеванию подвержены люди любой расовой принадлежности. У членов одной семьи нередко встречаются разные фенотипы. Ожидается, что с увеличением охвата новорожденных с помощью неонатального скрининга в США и др. странах точность оценки показателей заболеваемости повысится.
3. Патология. При электронной микроскопии клеток коры надпочечников, клеток Лейдига яичек и макрофагов НС можно обнаружить характерные ламеллярные включения. Эти включения, вероятно, состоят из ХС, эстерифицированного ОДЦЖК. Больше всего таких включений в клетках пучковой зоны коры надпочечников, которые сначала переполняются от липидов, а затем подвергаются атрофии.
В НС при АЛД наблюдают два типа патологических очагов. При тяжелой церебральной форме демиелинизация сопровождается воспалительным ответом в виде периваскулярных скоплений лимфоцитов, которые наиболее выражены в области, вовлеченной в патологический процесс. При медленнопрогрессирующей взрослой форме, адреномиелоневропатии, основным признаком является дистальная аксонопатия длинных проводящих путей спинного мозга. При этой форме воспалительные изменения выражены слабо или отсутствуют.
4. Патогенез. Дисфункция надпочечников, вероятно, является прямым следствием накопления ОДЦЖК. Клетки пучковой зоны коры надпочечников переполняются от патологического накопления липидов. ХС, эстерифицированный ОДЦЖК, относительно устойчивы к действию гидролаз эфиров ХС, стимулируемых АКТЕ В связи с этим способность этих ферментов превращать ХС в активные стероиды ограничена. Кроме того, С способствуют избыточному повышению вязкости плазматической мембраны, что может приводить к нарушению функции рецепторов и др. клеточных функций.
Корреляция между неврологическим фенотипом и природой мутации или выраженностью биохим. дефекта, оцениваемой по уровням ОДЦЖК плазмы, или между степенью вовлечения надпочечников и степенью вовлечения НС, отсутствует. Тяжесть заболевания и скорость прогрессирования коррелируют с интенсивностью воспалительного ответа. Воспалительный ответ м.б. отчасти опосредован цитокинами. Кроме того, он может включать аутоиммунный ответ, который запускается избыточным количеством ОДЦЖК посредством неизвестного механизма. По-видимому, повреждение митохондрий и окислительный стресс также вносят свой вклад. Примерно у половины пациентов воспалительного ответа не наблюдается. Объяснений этому не найдено.
5. Клинические проявления. Существует пять относительно отдельных фенотипов АЛД, три из которых проявляются объективными и субъективными симптомами в детском возрасте. При всех фенотипах в первые 3-4 года жизни дети, как правило, развиваются нормально.
При детской церебральной форме АЛД первые симптомы чаще всего замечают в возрасте 4-8 лет. К наиболее распространенным начальным проявлениям относятся гиперактивность, невнимательность и ухудшение показателей школьной успеваемости у ребенка, который до этого учился хорошо. Во многих случаях отмечают нарушение слуховой различительной способности при сохраненном восприятии тонов. Это подтверждается трудностями с использованием телефона и значительно сниженными результатами тестов на интеллект, задания в которых нужно воспринимать на слух. Часто нарушено восприятие пространства. Кроме того, к начальным симптомам относятся зрительные нарушения, атаксия, плохой почерк, судороги и косоглазие.
Во многих случаях зрительные нарушения связаны скорее с вовлечением теменно-затылочной коры, чем с патологией глазных яблок или зрительного тракта. По этой причине зрительные возможности страдают в разной степени и кажутся непостоянными. Судороги возникают почти у всех пациентов и м.б. первым проявлением заболевания. У некоторых пациентов повышено ВЧД. У 85% пациентов нарушено высвобождение кортизола в ответ на стимуляцию АКТЕ и отмечается легкая гиперпигментация. Поскольку у большинства пациентов с таким фенотипом имеются церебральные симптомы, дисфункцию надпочечников у них распознают только после установления диагноза. Для церебральной детской формы АЛД характерна тенденция к быстрому прогрессированию с нарастанием спастичности и паралича, потерей зрения и слуха, а также способности говорить или глотать.
В среднем интервал между первым неврологическим симптомом и кажущимся вегетативным состоянием составляет 1,9 года. Пациенты могут жить в таком кажущемся вегетативном состоянии >10 лет.
При подростковой форме АЛД неврологические симптомы возникают у пациентов в возрасте от 10 лет до 21 года. Проявления сходны с проявлениями детской церебральной формы АЛД, но при подростковой форме заболевание прогрессирует более медленно. У 10% пациентов заболевание проявляется в острой форме эпилептическим статусом, надпочечниковым кризом, острой энцефалопатией или комой.
Адреномиелоневропатия впервые проявляется в старшем подростковом или взрослом возрасте прогрессирующим парапарезом, обусловленным дегенерацией длинных проводящих путей спинного мозга. Примерно у половины пациентов мужского пола в патологический процесс вовлекается белое в-во ГМ.
Большое значение имеет фенотип, проявляющийся симптомами болезни Аддисона. У 25% пациентов мужского пола с болезнью Аддисона м.б. биохим. дефекты, связанные с АЛД. У многих из них поражение НС может отсутствовать, тогда как у др. могут наблюдаться легкие неврологические симптомы. Адреномиелоневропатия у многих развивается только во взрослом возрасте.
Термин «бессимптомная АЛД» относится к людям, у которых имеются биохимические отклонения, характерные для АЛД, но отсутствуют неврологические или эндокринные нарушения. Почти у всех людей, имеющих генетический дефект, в конечном счете появляются неврологические симптомы.
У 50% гетерозиготных женщин развивается синдром, напоминающий адреномиелоневропатию, но с менее тяжелыми проявлениями и более поздним началом. Надпочечниковая недостаточность и поражение ГМ развиваются редко.
У родственников пациентов, страдающих адреномиелоневропатией, наблюдают случаи типичной АЛД. Одна из наиболее сложных проблем в лечении АЛД заключается в том, что во многих случаях у пациентов, являющихся членами одной семьи, заболевание протекает совершенно по-разному. Напр., в одной и той же семье у мальчика может наблюдаться классическая форма АЛД с тяжелым течением, приведшая к смертельному исходу в возрасте 10 лет, а у его брата — адреномиелоневропатия с поздним началом.
6. Лабораторные и рентгенологические признаки. Наиболее специфическим и значимым лабораторным признаком являются высокие уровни ОДЦЖК в плазме крови, эритроцитах или в культуре кожных фибробластов. Положительные результаты получают у всех пациентов мужского пола с АЛД и у 85% носителей АЛД женского пола. Самым надежным методом выявления носителей является мутационный анализ. Для установления диагноза АЛД простого обнаружения вариации гена ABCD1 недостаточно. Необходимо выделить варианты, сопровождающиеся повышением уровней ОДЦЖК.
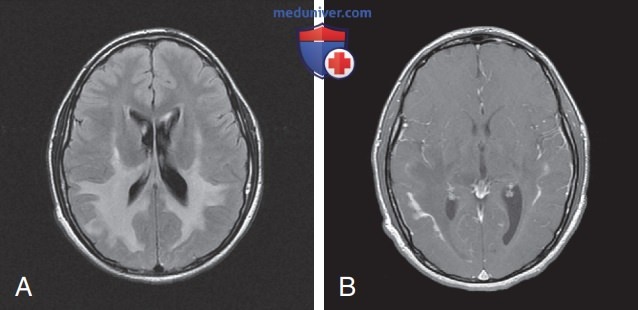
- Нейровизуализация. При МРТ у пациентов с детской церебральной или подростковой формой АЛД обнаруживаются характерные очаги в белом в-ве. У 80% пациентов очаги симметричны и обнаруживаются в валике мозолистого тела и перивентрикулярном белом в-ве теменной и затылочной долей. У многих пациентов имеются очаги накопления контраста в виде гирлянды кпереди от гипоэхогенных очагов, расположенных в задних отделах ГМ (рис. 5). Эта зона соответствует зонам интенсивной периваскулярной лимфоцитарной инфильтрации, в которой нарушен ГЭБ. У 10-15% пациентов первые очаги появляются в лобной доле. В редких случаях могут возникать односторонние очаги, которые благодаря масс-эффекту похожи на опухоль ГМ.
Рисунок 5. Характерные MPT-признаки церебральной формы адренолейкодистрофии: А — симметричные патологические изменения на Т2-взвешенных MPT-изображениях в белом в-ве задних отделов ГМ с вовлечением мозолистого тела; В — после введения контрастирующего в-ва обнаруживаются очаги усиления контраста в виде гирлянды
МРТ позволяет получить более четкое изображение нормального и патологически измененного белого в-ва, чем КТ, и является более предпочтительным методом визуализации.
- Нарушение функции надпочечников. Более чем у 85% пациентов с детской формой АЛД определяются повышенные уровни АКТГ плазмы и снижение уровня кортизола плазмы после в/в инъекции 250 мкг АКТГ (тетракозактида).
7. Постановка диагноза и проведение дифференциальной диагностики. Благодаря неонатальному скринингу, который добавили к единой скрининговой панели, стало возможным установление диагноза у бессимптомных пациентов мужского пола. После установления диагноза следует провести подтверждающее исследование и генетическое консультирование. Затем эти пациенты мужского пола вступают в программу наблюдения за надпочечниковой недостаточностью и раннего выявления возможного заболевания ГМ. Лица женского пола, выявленные с помощью указанной программы, также должны пройти подтверждающие исследования, генетическое консультирование семьи и скрининговое обследование др. заболеваний лицам мужского пола из группы риска. Представительницам женского пола в детском возрасте, как правило, не требуется никакого контрольного обследования, кроме вышеуказанного.
Самые ранние клинические проявления детской церебральной формы АЛД сложно отличить от более распространенных расстройств, сопровождающихся дефицитом внимания или снижением способности к обучению в школьном возрасте. На АЛД указывают быстрое прогрессирование болезни, признаки деменции или нарушение восприятия речи. Даже на ранних стадиях можно обнаружить патологические изменения с помощью методов нейровизуализации. В некоторых случаях др. лейкодистрофии или рассеянный склероз могут сопровождаться такими же изменениями, однако АЛД на ранних стадиях характеризуется большей склонностью к расположению очагов в задних отделах ГМ, чем указанные заболевания. Окончательный диагноз выставляют на основании избытка ОДЦЖК, который возникает только при АЛД и др. пероксисомных нарушениях.
Церебральные формы АЛД, особенно асимметричные, можно спутать с глиомами или др. объемными образованиями. До установления правильного диагноза пациентам выполняют биопсию ГМ и, в редких случаях, проводят лечение. Наиболее надежным методом, позволяющим провести ДД, является определение уровней ОДЦЖК в плазме крови.
У подростков или взрослых церебральную форму АЛД можно ошибочно принять за психиатрические нарушения, деменцию, рассеянный склероз или эпилепсию. Первым признаком диагноза АЛД м.б. обнаружение характерных очагов в белом в-ве ГМ при проведении нейровизуализационных методов обследования. Определение уровней ОДЦЖК подтверждает диагноз.
АЛД невозможно отличить от др. форм болезни Аддисона по клиническим признакам. Всем пациентам мужского пола с болезнью Аддисона рекомендуется проводить определение уровней ОДЦЖК. В плазме крови пациентов с АЛД, как правило, отсутствуют АТл к ткани надпочечников.
8. Осложнения. К предотвратимым осложнениям относится надпочечниковая недостаточность. Труднее всего бороться с неврологическими осложнениями, которые появляются у пациентов, прикованных к постели. Это контрактуры, кома и нарушение глотания. Др. осложнения включают поведенческие нарушения и травмы, связанные с нарушением ориентации в пространстве, нарушением зрения и слуха, а также с судорожными припадками.
9. Лечение. При надпочечниковой недостаточности или гипофункции коры надпочечников эффективна заместительная терапия ГКС. Такая терапия может сохранить жизнь пациенту, поддержать силы и улучшить самочувствие, однако она не влияет на общее течение инвалидизирующих неврологических осложнений.
- Трансплантация костного мозга. Трансплантация костного мозга (ТКМ) или гемопоэтических стволовых клеток эффективна у пациентов с ранними признаками воспалительной демиелинизации, которая сопровождается быстрым прогрессированием инвалидизирующих неврологических нарушений и характерна для мальчиков и подростков с фенотипом церебральной формы АЛД. ТКМ — рискованная процедура, требующая тщательного отбора и обследования пациентов. Механизм ее положительного эффекта до конца не ясен.
Клетки костномозгового происхождения экспрессируют пероксисомный мембранный белок АЛД, которого недостаточно при АЛД. Примерно 50% клеток микроглии ГМ имеют костномозговое происхождение. Благоприятное воздействие ТКМ м.б. связано с модификацией воспалительного ответа в ГМ. Последующее наблюдение мальчиков и подростков, у которых имелись начальные патологические изменения ГМ, показало, что процесс стабилизировался. С др. стороны, при тяжелых изменениях ГМ ТКМ не останавливает процесс и может ускорить прогрессирование заболевания.
Результаты балльной оценки изменений на MPT-изображениях и результаты тестов на IQ при АЛД позволяют в некоторой степени спрогнозировать эффективность ТКМ у мальчиков. При результатах тестов на IQ <80 баллов проводить трансплантацию не рекомендуют. К сожалению, более чем у половины пациентов, у которых поводом для обследования становятся неврологические симптомы, заболевание на данный момент настолько прогрессировало, что им не показана трансплантация.
ТКМ наиболее целесообразно проводить при отсутствии неврологических симптомов или при незначительном вовлечении НС в патологический процесс. Чаще всего таких пациентов выявляют при скрининговом обследовании относящихся к группе риска родственников пациентов, у которых имеются клинические проявления. Кроме того, кандидатов на ТКМ можно определить при скрининговом обследовании уровней ОДЦЖК плазмы у пациентов с болезнью Аддисона. ТКМ сопряжена с высоким риском для жизни (летальность составляет 10-20%). Поскольку у 50% нелеченых пациентов с АЛД воспалительная демиелинизация ГМ не развивается, трансплантацию не рекомендуют пациентам, у которых по данным МРТ нет поражения ГМ. Патологические изменения на MPT-изображениях возникают раньше явных неврологических симптомов или нейропсихологических отклонений.
У мальчиков и подростков 3-15 лет, не имеющих неврологической симптоматики, МРТ следует проводить каждые 6 мес. При отсутствии патологических изменений по данным МРТ проведение ТКМ не показано. В случае развития патологических изменений по результатам МРТ мальчиков обследуют в центре, владеющем методикой проведения трансплантации у пациентов с АЛД. Обследование должно включать неврологическое и нейропсихологическое исследования, а также МРТ. Оказывает ли ТКМ положительное влияние у взрослых с фенотипом адреномиелоневропатии и невоспалительными изменениями в спинном мозге, неизвестно.
- Лечение маслом Лоренцо. На данный момент исследуют эффективность применения масла Лоренцо (смесь глицеролтриолеата и глицеролтриэруката 4:1) в сочетании с диетой в отношении профилактики развития различных проявлений АЛД. Указанное соединение действительно снижает уровни ОДЦЖК плазмы, но, несмотря на первоначальный «+» результат, клинические исследования неоднозначны. Не было продемонстрировано, что применение масла Лоренцо влияет на прогрессирование заболевания у пациентов мужского пола с церебральной формой АЛД. Пока неясно, влияет ли этот или др. ЛП, снижающий уровни ОДЦЖК, на течение заболевания.
- Поддерживающая терапия. Прогрессирующие поведенческие и неврологические нарушения, которые развиваются на фоне детской формы АЛД, представляют огромную трудность для семьи. Пациентам с АЛД требуется полноценная программа лечения и партнерство между семьей, врачом, медсестрами, школьной администрацией, учителями и др. вспомогательным персоналом. Кроме того, нередко помогают родительские группы поддержки (напр., Объединенный фонд лейкодистрофии). Большое значение имеет взаимодействие со школьной администрацией, поскольку, согласно положениям США (публичный закон 94-142) дети с ALD имеют право на получение специальных услуг по категории «др. нарушения здоровья» или «несколько видов инвалидности».
В зависимости от скорости прогрессирования заболевания особые потребности могут варьировать от относительно низкозатратных услуг в рамках обычной школьной программы до домашних или больничных программ обучения для немобильных детей.
Трудности лечения варьируют в зависимости от стадии заболевания. Ранние стадии характеризуются незначительными изменениями в сфере эмоций, поведения и внимания. В этот период основное значение имеют консультации со специалистами и взаимодействие со школьной администрацией и учителями. Изменения в цикле «сон-бодрствование» можно скорректировать разумным применением ночных снотворных ЛС.
С прогрессированием лейкодистрофии основными проблемами становятся изменения тонуса мышц и поддержка функции мышц, иннервируемых бульбарной группой нервов. Эффективным ЛС для лечения острых эпизодических болезненных мышечных спазмов является баклофен в постепенно повышающихся дозах (от 5 мг 2 р/сут до 25 мг 4 р/сут). Можно также использовать др. ЛП. При этом необходимо тщательно отслеживать побочные эффекты и лекарственные взаимодействия. По мере прогрессирования лейкодистрофии теряется контроль над бульбарными мышцами. В первое время проблему можно решить изменением диеты в сторону кашицеобразной пищи, однако в конечном счете большинство пациентов нуждаются в назогастральном зонде или установке гастростомы. По меньшей мере у 30% пациентов возникают фокальные или генерализованные судорожные припадки, которые, как правило, хорошо отвечают на терапию стандартными противосудорожными ЛП.
10. Генетическое консультирование и профилактика. Генетическое консультирование и соответствующий мониторинг имеют ключевое значение. Всем входящим в группу риска родственникам пациентов, имеющих клинические проявления, нужно проводить скрининговое обследование. Благодаря одной скрининговой программе удалось выявить >250 бессимптомных пораженных представителей мужского пола и 1200 девочек и женщин, гетерозиготных по АЛД. Исследование плазмы крови позволяет с высокой степенью достоверности обнаружить пораженных мальчиков и мужчин, у которых уровни ОДЦЖК плазмы повышены уже на момент рождения. Выявление бессимптомных мальчиков и мужчин позволяет своевременно начать заместительную терапию стероидами и предотвратить надпочечниковый криз, который может привести к летальному исходу.
Проведение повторных MPT-исследований ГМ также позволяет выявить среди пациентов кандидатов на ТКМ на той стадии, когда эта процедура имеет максимальные шансы на успех. Кроме того, уровни ОДЦЖК плазмы рекомендуется определять всем пациентам мужского пола с болезнью Аддисона. Показано, что в >25% случаев надпочечниковой недостаточности неизвестной этиологии у мальчиков причиной является АЛД. Женщин, гетерозиготных по АЛД, выявить намного сложнее, чем пораженных мальчиков и мужчин. У 15-20% гетерозиготных женщин уровни ОДЦЖК плазмы не отклоняются от нормы, и то, что на этот факт не обращали внимания, привело к серьезным ошибкам при генетическом консультировании. Анализ ДНК позволяет с высокой степенью достоверности идентифицировать носителей при условии определения мутации у члена семьи. Эту процедуру рекомендуется проводить с целью выявления гетерозиготных женщин.
У пораженных плодов мужского пола диагноз можно установить пренатально путем определения известной мутации или измерения уровней ОДЦЖК в культуре амниоцитов или в клетках ворсинок хориона. По возможности при выявлении нового пациента с АЛД следует составить подробную родословную и попытаться обнаружить всех входящих в группу риска носителей женского пола и пораженных мужчин. Проводя указанные исследования при консультировании, необходимо проявлять сочувствие и уделять внимание социальной, эмоциональной и этической сторонам проблемы.