Глутаминовая кислота и ее амидное производное глутамин выполняют в организме широкий спектр функций. Глутамат осуществляет многочисленные биологические функции, действуя как нейромедиатор, промежуточное соединение во многих фундаментальных биохим. реакциях и предшественник ингибирующего нейромедиатора ГАМК.
Др. важным производным глутамата является глутатион (γ-глутамилцистеинилглицин). Синтез и разложение этого распространенного трипептида, выполняющего функцию основного антиоксиданта в организме, осуществляется в рамках сложного цикла, называемого γ-глутамиловым циклом (рис. ниже). Глутатион, благодаря наличию свободной сульфгидрильной (-SH) группы и своему высокому содержанию в клетках, защищает др. сульфгидрилсодержащие соединения (напр., ферменты, КоА) от окисления.
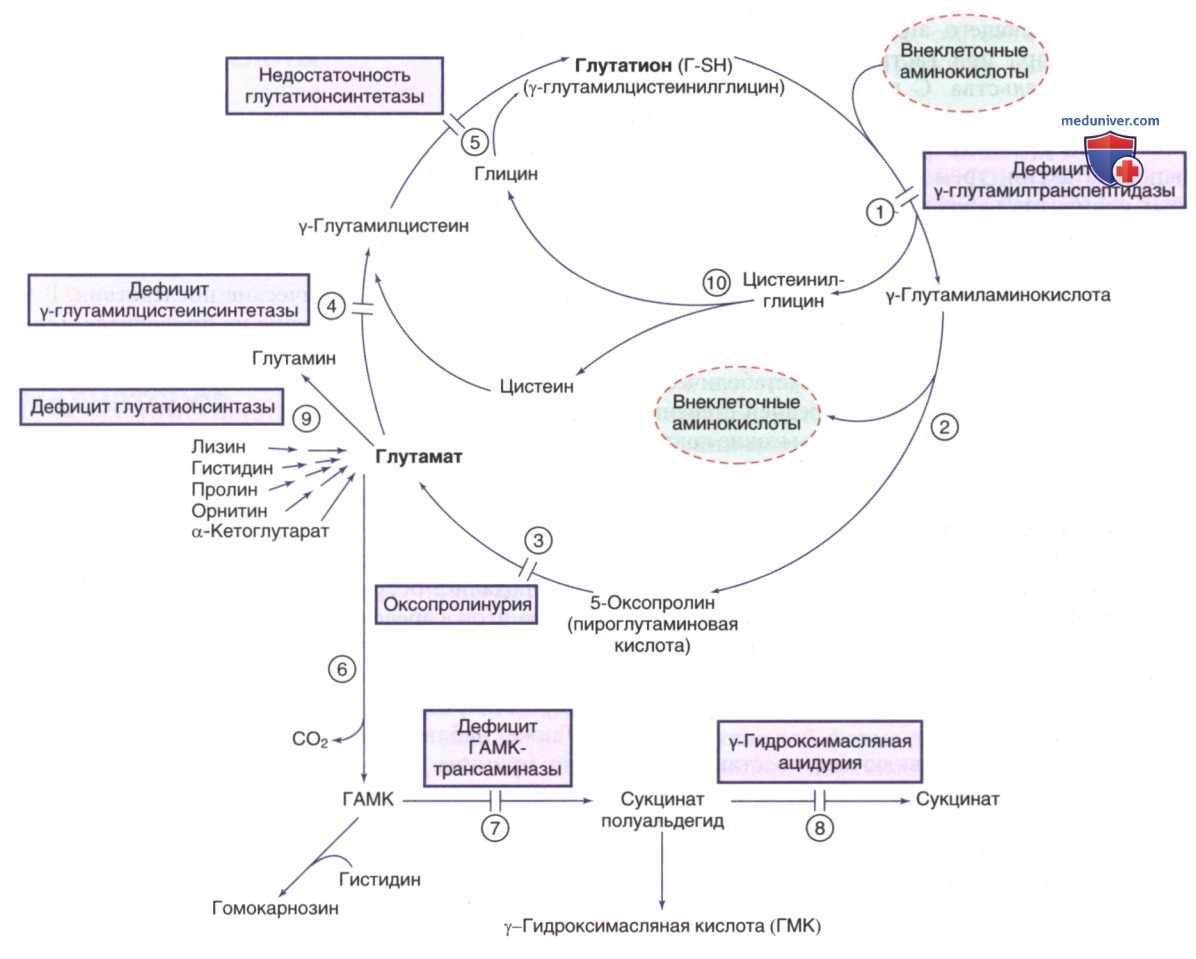
γ-Глутамиловый цикл и связанные с ним пути. Отмечены нарушения синтеза и распада глутатиона (Г-SH). Ферменты: 1) γ-глутамилтранспептидаза (ГГТ); 2) у-глутамилциклотрансфераза; 3) 5-оксопролиназа; 4) у-глутамилцистеинсинтетаза; 5) глутатионсинтетаза; 6) декарбоксилаза глутаминовой кислоты; 7) трансаминаза гамма-аминомасляной кислоты (ГАМК); 8) сукцинат-полуальдегиддегидрогеназа; 9) глутаминсинтетаза; 10) дипептидаза
Он также участвует в детоксикации пероксидов, в т.ч. перекиси водорода, и поддерживает восстановительный потенциал клеток. Кроме того, глутатион участвует в транспорте аминокислот через клеточную мембрану посредством у-глутамилового цикла.
Одним из биохим. проявлений дефицита γ-глутамилового цикла является повышенная экскреция 5-оксопролина с мочой, что м.б. результатом как генетических, так и негенетических причин. Необходимо проводить ДД 5-оксопролинемии с метаболическим ацидозом с высокой анионной разницей. Массивная 5-оксопролинурия может проявляться при двух метаболических нарушениях: дефицит глутатионсинтетазы и недостаточность 5-ок-сопролиназы (см. рис. выше).
Однако наиболее часто встречается временное и умеренное повышение уровня 5-оксопролина в моче, вызванное метаболическими нарушениями и приобретенными заболеваниями, напр. воздействием парацетамола («Ацетаминофена») и некоторых смесей с гидролизованным белком, тяжелыми ожогами, синдромом Стивенса-Джонсона (Stevens-Johnson), гомоцистинурией, нарушением цикла мочевины и тирозинемией типа I.
а) Дефицит глутатионсинтетазы. Зарегистрированы три формы этого редкого заболевания. В легкой форме дефицит ферментов приводит к дефициту глутатиона только в эритроцитах. У таких пациентов развивается гемолитическая анемия без хронического метаболического ацидоза и сохраняется высокая остаточная активность глутатионсинтетазы при ферментативном исследовании. При средней форме гемолитическая анемия сочетается с разл. степенью метаболического ацидоза и 5-оксопролинурией.
Тяжелая форма заболевания отличается наличием гемолитической анемии, сопровождающейся тяжелым ацидозом, массивной 5-оксопролинурией и неврологическими проявлениями.
1. Тяжелые и средние формы дефицита глутатионсинтетазы. У новорожденных с этой редкой патологией обычно развиваются симптомы острого метаболического ацидоза, желтухи, гемолитической анемии легкой и средней степени тяжести в первые несколько дней жизни. После выздоровления остается хронический ацидоз. Подобные эпизоды жизнеугрожающего ацидоза могут возникать при инфекциях (напр., при гастроэнтерите)/после хирургического вмешательства.
С возрастом развиваются прогрессирующие неврологические нарушения, которые проявляются в виде умственной отсталости, спастического тетрапареза, атаксии, тремора, дизартрии и судорог.
У некоторых пациентов наблюдается восприимчивость к инфекциям, предположительно из-за дисфункции гранулоцитов. Пациенты со средней формой дефицита глутатионсинтетазы имеют более легкий ацидоз и менее выраженную 5-оксопролинурию, чем при тяжелой форме, при этом неврологические проявления отсутствуют.
К лабораторным признакам относятся метаболический ацидоз, гемолитическая анемия легкой/средней степени и 5-оксопролинурия. В крови присутствуют высокие концентрации 5-оксопролина. У пациентов со средней тяжестью заболевания уровни 5-оксопролина в моче и крови ниже. Содержание глутатиона в эритроцитах заметно снижено. Повышенный синтез 5-оксопролина при этом заболевании считается результатом превращения γ-глутамилцистеина в 5-оксопролин ферментом γ-глутамилциклотрансферазой (см. рис. выше).
Продуцирование γ-глутамилцистеина значительно увеличивается, поскольку устраняется нормальное ингибирующее влияние глутатиона на фермент у-глутамилцистеинсинтетазу.
Лечение острого приступа включает восстановление водного баланса, коррекцию ацидоза [инфузионным введением натрия гидрокарбоната («Натрия бикарбонат»)] и меры по коррекции анемии и гипербилирубинемии. Ощелачивание организма приходится проводить постоянно. Рекомендуется принимать БАД с витамином С, витамином Е и селеном. Следует избегать приема ЛП и оксидантов, которые вызывают гемолиз, и предотвращать ситуации, сопровождающиеся усилением катаболизма. Прием внутрь аналогов глутатиона имеет переменный успех.
Пренатальную диагностику выполняют путем измерения 5-оксопролина в околоплодных водах, исследования активности фермента в культуре амниоцитов/образцах ворсинок хориона, а также ДНК-анализа. Зарегистрирован случай успешной беременности женщины с этой патологией (средняя форма) и благоприятным исходом как для матери, так и для ребенка.
2. Легкая форма недостаточности глутатионсинтетазы. Зарегистрировано лишь несколько пациентов с легкой формой этой патологии. Единственным клиническим признаком была гемолитическая анемия легкой/средней степени. У некоторых пациентов наблюдалась спленомегалия. Когнитивные функции не нарушены. Хронический метаболический ацидоз обычно не наблюдается. У некоторых пациентов м.б. повышена концентрация 5-оксопролина в моче.
Патогенные варианты гена этого фермента (GSSD), предположительно, уменьшают период полужизни фермента, что приводит к повышенной скорости обмена белка при сохранении его каталитической функции. Ускоренный распад фермента в этих случаях не сказывается на клетках, в которых происходит нормальный синтез белка. При этом неспособность зрелых эритроцитов синтезировать белок приводит к дефициту глутатиона в эритроцитах. Лечение аналогично лечению гемолитической анемии и заключается в отказе от ЛП и оксидантов, способных вызвать гемолиз.
Все формы дефицита глутатионсинтетазы наследуются по АуР-типу. GSSD расположен в хромосоме 20q11.22. Диагноз подтверждают путем ДНК-анализа/определения активности ферментов в эритроцитах и фибробластах кожи.
3. Дефицит 5-оксопролиназы. Описано >20 пациентов с 5-оксопролинурией (4-10 г/сут), вызванной дефицитом 5-оксопролиназы (см. рис. выше). Специфической клинической картины пока не отмечено; есть полностью бессимптомные пациенты. Поэтому неясно, имеет ли дефицит 5-оксопролиназы какие-либо клинические последствия. В настоящее время специальное лечение не проводится. Ген фермента (OPLAH) расположен в хромосоме 8q24.3.
4. Дефицит γ-глутамилцистеинсинтетазы (дефицит глутаматцистеинлигазы). Зарегистрировано только несколько пациентов с дефицитом данного фермента. Наиболее постоянным клиническим проявлением была хроническая гемолитическая анемия легкой степени. При воздействии сульфаниламидов наблюдались острые приступы гемолиза. У двух взрослых сиблингов в зрелом возрасте были отмечены периферическая невропатия и прогрессирующая спиномозжечковая дегенерация. У всех пациентов присутствовали лабораторные признаки хронической гемолитической анемии.
Также наблюдается генерализованная аминоацидурия по причине того, что γ-глутамиловый цикл участвует в транспорте аминокислот в клетках (см. рис. выше). Лечение должно быть сфокусировано на терапии гемолитической анемии и отказе от ЛП и оксидантов, способных вызвать гемолиз. Заболевание наследуется по АуР-типу; ген (GCLC) расположен в хромосоме 6p12.1.
б) Дефицит γ-глутамилтранспептидазы (глутатионемии). γ-Глутамилтранспептидаза присутствует в любой клетке, выполняющей секреторную/абсорбционную функцию. Ее особенно много в почках, ПЖЖ, кишечнике и печени. Фермент также присутствует в желчи. Измерение у-глутамилтранспептидазы в крови часто выполняется для диагностики заболеваний печени и желчных протоков.
Дефицит фермента приводит к повышению концентрации глутатиона в физиол. жидкостях, но при этом его концентрация в клетках остается нормальной (см. рис. выше). Зарегистрировано всего несколько пациентов с дефицитом γ-глутамилтранспептидазы, поэтому клинические проявления такой патологии охарактеризовать трудно. У трех пациентов наблюдалась умственная отсталость легкой/средней степени и серьезные поведенческие проблемы. Однако одна из двух сестер с этим заболеванием отличалась нормальным интеллектом во взрослом возрасте, у другой же был диагностирован синдром Прадера-Вилли.
К лабораторным признакам относятся заметное повышение концентрации глутатиона (до 1 г/сут), γ-глутамилцистеина и цистеина в моче. Ни у одного из зарегистрированных пациентов не наблюдалось генерализованной аминоацидурии (ожидаемый признак при дефиците этого фермента) (см. рис. выше).
Диагноз подтверждается путем измерения активности фермента в лейкоцитах/культуре фибробластов кожи. Эффективного лечения в настоящее время не существует. Заболевание наследуется как очевидный АуР-признак. γ-Глутамилтранспептидазы представляют собой большое семейство ферментов, кодируемых по меньшей мере семью генами.
в) Генетические нарушения метаболизма γ-аминомасляной кислоты. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.