а) Нарушение обмена гистидина. Гистидин разлагается по пути урокановой кислоты до глутаминовой кислоты. Зарегистрировано несколько генетических биохим. отклонений, связанных с процессом разложения гистидина, но клиническое значение повышенных уровней гистидина не установлено.
Декарбоксилирование гистидина гистидиндекарбоксилазой приводит к образованию гистамина. Дефицит этого фермента обусловливает развитие семейной формы синдрома Туретта.
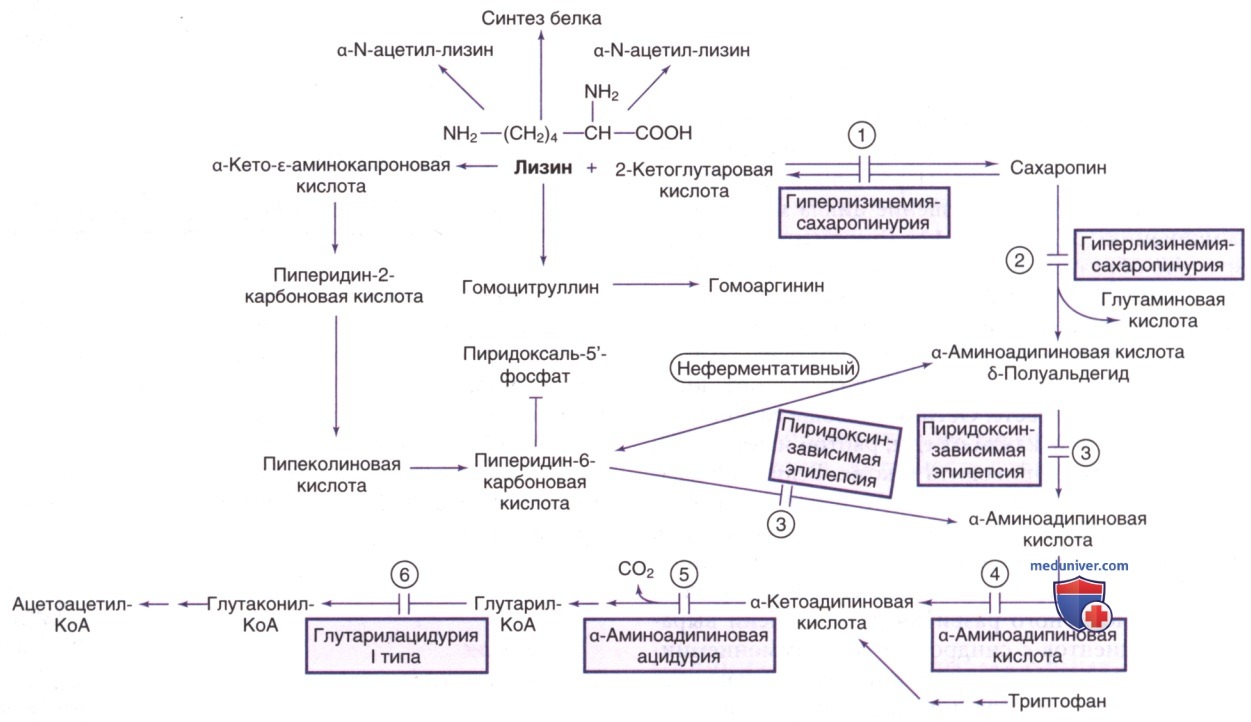
б) Нарушение обмена лизина. Лизин катаболизируется двумя путями. В первом пути лизин конденсируется с α-кетоглутаровой кислотой с образованием сахаропина. Затем сахаропин катаболизируется до α-аминоадипинового полуальдегида и глутаминовой кислоты. Эти первые две стадии катализируются синтетазой α-аминоадипинового полуальдегида, которая имеет два активных центра: лизинкетоглутаратредуктазу и сахаропиндегидрогеназу (рис. 14).
Во втором пути лизин сначала трансаминируется, а затем конденсируется до своих циклических форм (пипеколиновой кислоты и пиперидин-6-карбоновой кислоты). Пиперидин-6-карбоновая кислота и ее линейная форма (α-аминоадипиновый полуальдегид) окисляются до α-аминоадипиновой кислоты ферментом антиквитином. Это основной путь D-лизина в организме и L-лизина в ГМ.
Гиперлизинемия-сахаропинурия и α-аминоадип-α-кетоадипиновая ацидемия являются биохим. заболеваниями, вызванными врожденными нарушениями разложения лизина. Эти заболевания обычно протекают бессимптомно.
в) Пиридоксин (витамин В6-зависимая) эпилепсия. Пиридоксаль-5’-фосфат, активная форма пиридоксина, является кофактором многих ферментов, в т.ч. участвующих в метаболизме нейромедиаторов. В/клеточный дефицит пиридоксаль-5’-фосфата в ГМ может привести к судорожному расстройству, невосприимчивому к обычным противосудорожным ЛП, но отвечающему на высокие дозы пиридоксина. Эти пиридоксин-зависимые фенотипы наблюдаются при следующих генетических метаболических нарушениях.
1. Дефицит антиквитина (дегидрогеназы α-аминоадипинового полуальдегида). Это наиболее частая причина пиридоксин-зави-симой эпилепсии. Дефицит антиквитина приводит к накоплению пиперидин-6-карбоновой кислоты в ткани ГМ (см. рис. 14); она реагирует с пиридоксаль-5’-фосфатом и делает его неактивным. Т.о., для преодоления этой инактивации необходимы большие дозы пиридоксина. Состояние наследуется по АуР-типу; ген антиквитина (ALDH7A1) расположен в хромосоме 5q31.
2. Дефицит пиридокс(ам)ин-5'-фосфатоксидазы. Клинические проявления дефицита пиридокс(ам)ин-5’-фосфатоксидазы совпадают с дефицитом антиквитина. У пациентов часто наблюдаются судороги, начинающиеся в неонатальном периоде, задержка психического развития, спастическая тетраплегия и неспецифические признаки при визуализации ГМ (отсроченная миелинизация, церебральная атрофия и аномальные сигналы в базальных ганглиях).
Зарегистрировано не так много случаев регресса психического развития, бледности диска зрительного нерва и ретинопатии. Аминокислотный анализ плазмы и СМЖ может выявить повышенный уровень глицина, при котором потребуется обследование на НКГ и более позднее начало лечения пиридоксаль-5’-фосфатом. Анализ нейромедиаторов в СМЖ выявил несоответствующие изменения концентраций 3-О-метилдофа, гомованилиновой кислоты и 5-гидроксииндолеуксусной кислоты. У одного пациента был зарегистрирован нормальный уровень пиридоксаль-5’-фосфата в СМЖ, что позволяет предположить, что пробное лечение пиридоксаль-5’-фосфатом и молекулярный анализ м.б. разумной стратегией у некоторых пациентов независимо от результатов анализа СМЖ.
Для недопущения токсического эффекта следует использовать самую низкую эффективную дозу пиридоксаль-5’-фосфатом. Нарушение вызвано АуР-патогенными вариантами PNPO.
3. Дефицит сульфитоксидазы и дефицит кофактора молибдена. При этом редком заболевании накопление сульфитов приводит к ингибированию ферментативной активности антиквитина и накоплению пиперидин-6-карбоновой кислоты, что, в свою очередь, вызывает инактивацию пиридоксаль-5’-фосфата и зависимость от витамина В6.
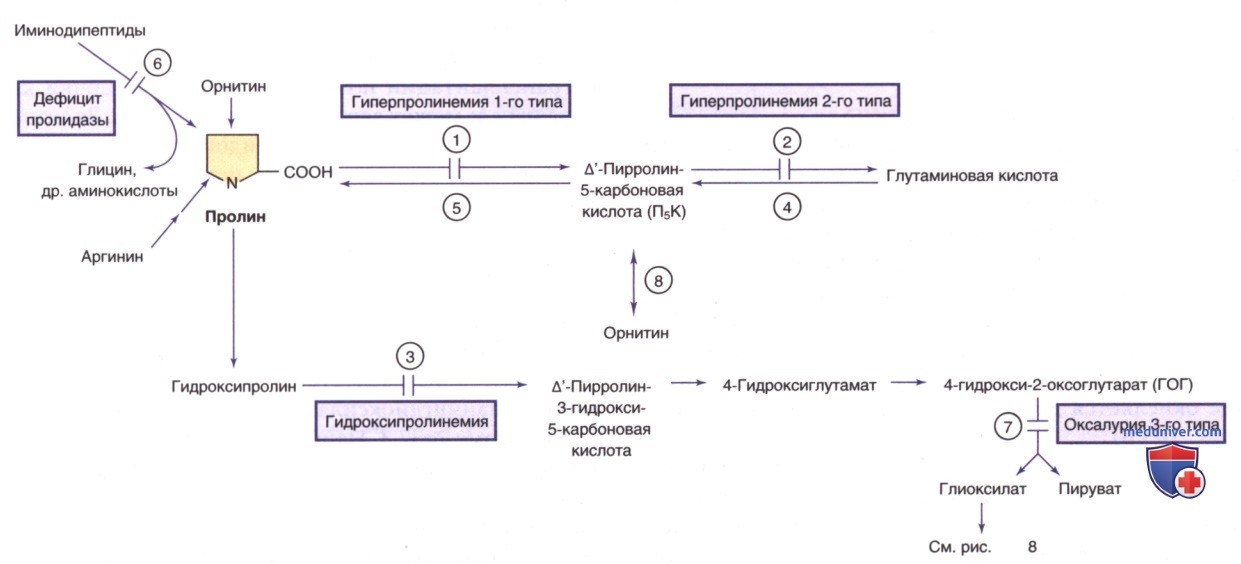
4. Гиперпролинемия типа II. При этом заболевании накопление А1-пирролин-5-карбоксилата в ткани ГМ вызывает инактивацию пиридоксаль-5’-фосфата, что приводит к зависимости от пиридоксина (рис. 9).
5. Гипофосфатазия. Пиридоксаль-5’-фосфат является основной циркулирующей формой пиридоксина. Для дефосфорилирования пиридоксаль-5’-фосфата необходима ЩФ. При этом образуется свободный пиридоксин, который является единственной формой витамина В6, способной преодолевать ГЭБ и проникать в клетки ГМ. Рефосфорилирование пиридоксина протекает в/клеточно с образованием пиридоксаль-5’-фосфата. При неонатальной форме гипофосфатазии пиридоксаль-5’-фосфат не может дефосфорилироваться до свободного пиридоксина из-за выраженного дефицита тканенеспецифической ЩФ. Это приводит к дефициту пиридоксина в ГМ и развитию пиридоксинзависимой эпилепсии.
Основным клиническим проявлением пиридоксин-зависимой эпилепсии, вызванной дефицитом антиквитина, являются генерализованные судороги, которые обычно возникают в первые дни жизни и не поддаются лечению обычными противосудорожными ЛП. Некоторые матери во время беременности отмечают аномальное в/утробное дрожание у ребенка с такой патологией. Судороги обычно тонико-клонические, но м.б. практически любого типа. Могут наблюдаться др. проявления, такие как дистония, нарушения дыхания, вздутие живота и рвота, гепатомегалия, гипогликемия и гипотермия. Частыми последствиями являются проблемы с обучением и задержка речи.
Также зарегистрированы формы заболевания с более поздним началом (до 5 лет). Следовательно, любому новорожденному с труднокупируемыми судорогами рекомендуется проводить анализ на витамин В6.
К лабораторным признакам относятся повышенные концентрации а-аминоадипинового полуальдегида и пипеколиновой кислоты в СМЖ, плазме и моче. После лечения отклонения ЭЭГ могут нормализоваться. Результаты нейровизуализирующих исследований м.б. в норме, однако сообщалось о церебральной атрофии и атрофии мозжечка, перивентрикулярной гиперинтенсивности, в/мозговых кровоизлияниях и гидроцефалии.
Лечение витамином В6 (50-100 мг/сут) обычно приводит к резкому уменьшению судорог и аномалий ЭЭГ. Высокие дозы пиридоксина могут привести к развитию периферической невропатии, поэтому следует избегать доз >500 мг/сут. Зависимость от пиридоксина и, следовательно, терапия сохраняются на протяжении всей жизни. В настоящее время проводится оценка эффективности диеты с ограничением лизина.
г) Глутаровая ацидурия 1-го типа (дефицит глутарил-КоА-дегидрогеназы). Глутаровая кислота является промежуточным соединением при распаде лизина (см. рис. 14), гидроксилизина и триптофана. Глутаровую ацидурию 1-го типа (нарушение, вызванное дефицитом глутарил-КоА-дегидрогеназы) следует отличать от глутаровой ацидурии 2-го типа (отдельного клинического и биохим. нарушения, вызванного дефектами в митохондриальной цепи транспорта электронов).
1. Клинические проявления. Макроцефалия является частым, но неспецифическим проявлением глутаровой ацидурии 1-го типа. Она развивается в 1-й год жизни, но может присутствовать уже при рождении и предшествовать появлению неврологических симптомов. У некоторых младенцев с этой патологией могут проявляться слабовыраженные неврологические симптомы, такие как задержка моторного развития, повышенная возбудимость и проблемы с кормлением в период, кажущийся бессимптомным. Начало заболевания обычно отмечается острыми энцефалопатическими признаками, такими как потеря нормальных моторных навыков (ребенок перестает держать голову, переворачиваться и сидеть), судорогами, общей ригидностью, опистотонусом, хореоатетозом и дистонией, вызванными острым повреждением полосатого тела. Эти симптомы могут возникать внезапно после незначительной инфекции у внешне здорового ребенка.
Визуализирующие исследования ГМ выявляют увеличение экстрааксиальной (особенно фронтальной) жидкости с растянутыми переходными венами, поражениями полосатого тела, расширенными боковыми желудочками, кортикальной атрофией (в основном в лобновисочной области) и фиброзом. Восстановление после первого приступа обычно происходит медленно, при этом могут сохраняться некоторые остаточные неврологические нарушения, в частности дистония и хореоатетоз. При отсутствии лечения могут возникать новые острые приступы, похожие на первые, во время последующих эпизодов интеркуррентных инфекций/катаболических состояний.
У некоторых пациентов эти признаки и симптомы могут развиваться постепенно в первые несколько лет жизни. Гипотония и хореоатетоз могут постепенно прогрессировать до ригидности и дистонии (скрытая форма). При этой форме после инфекции/др. катаболических состояний развиваются острые эпизоды метаболической декомпенсации, рвота, кетоз, судороги и кома. При отсутствии лечения смерть может наступить <10 лет во время одного из этих эпизодов. Новорожденные с этой патологией склонны к формированию субдуральной гематомы и ретинального кровоизлияния после незначительных падений и травм головы. Эти симптомы можно ошибочно принять за жестокое обращение с детьми. У большинства пациентов интеллектуальные способности обычно остаются относительно нормальными.
2. Лабораторные признаки. Во время острых приступов может развиться метаболический ацидоз и кетоз легкой/средней степени. У некоторых пациентов наблюдаются гипогликемия, гипераммониемия и повышение уровня трансаминаз в сыворотке. В моче, крови и СМЖ обычно наблюдаются высокие концентрации глутаровой кислоты. Также в физиол. жидкостях может присутствовать 3-гидроксиглутаровая кислота. Профиль ацилкарнитина показывает повышенное содержание глутарилкарнитина (C5-DC) в крови и моче. Концентрации аминокислот в плазме обычно соответствуют норме.
В периоды между приступами лабораторные признаки могут не иметь значимых изменений. Глутаровую ацидурию 1-го типа можно определить при скрининге новорожденных путем измерения концентрации глутарилкарнитина в каплях крови. Чувствительность этого скринингового метода зависит от порогового значения, используемого программой скрининга новорожденных, и некоторые пациенты могут оказаться невыявленными. Напр., это может касаться подгруппы пациентов с глутаровой ацидурией 1-го типа с нормальным содержанием глутаровой кислоты в плазме и моче и несколько повышенным уровнем глутарилкарнитина в плазме. Этот тип глутаровой ацидурии 1-го типа, относящийся к фенотипу «с низкой экскрецией», несет такой же риск развития повреждения ГМ, как и при фенотипе «с высокой экскрецией». У некоторых пациентов с фенотипом низкой экскреции глутаровая кислота повышается только в СМЖ.
Определение глутарилкарнитина в моче, вероятно, является более чувствительным методом скрининга для выявления больных с низкой экскрецией. Любому ребенку с прогрессирующей дистонией и дискинезией необходимо провести анализ активности фермента глутарил-КоА-дегидрогеназы и молекулярный анализ GCDH.
3. Лечение. Пациентам необходимо соблюдать диету с ограничением лизина и триптофана, при этом обеспечиваются физиол. потребности в белке, питательных микроэлементах и витаминах. Повышенное содержание аргинина в пище позволяет снизить поглощение лизина клетками и уменьшить эндогенное образование глутарил-КоА. Пациентов следует регулярно обследовать на дефицит лизина и триптофана путем мониторинга аминокислот в плазме и измерения роста. Во всех случаях рекомендуется прием L-карнитина (внутрь 50-100 мг/кг в сутки). Для снижения риска повреждения полосатого тела важнейшее значение имеет незамедлительное начало лечения в период острого заболевания, в т.ч. временное прекращение приема белка на 24 ч, восполнение потерянных калорий с помощью углеводов/липидов, в/в введение L-карнитина и декстрозы («Глюкозы»), быстрое лечение инфекции и контроль лихорадки. Всем пациентам следует предоставить информацию по первой помощи с описанием основного диагноза и рекомендациями по обследованию и лечению.
Ранняя диагностика при скрининге новорожденных с профилактикой и интенсивным лечением интеркуррентных катаболических состояний (инфекций) позволяет минимизировать повреждение полосатого тела и обеспечить благоприятный прогноз. Пациентам с двигательным расстройством и спастичностью может потребоваться лечение баклофеном, диазепамом, тригексифенидилом и инъекциями ботулинического токсина типа А.
Глутаровая ацидурия 1-го типа наследуется как АуР-признак. По оценкам, частота встречаемости составляет 1:100 000 живорождений во всем мире. Заболевание чаще встречается в некоторых этнических группах (канадские индейцы оджи-кри, ирландские путешественники, чернокожие южноафриканцы, шведы и амиши старого порядка в США). Ген глутарил-КоА-дегидрогеназы (GCDH) расположен в хромосоме 19р13.2. Молекулярный анализ GCDH позволяет идентифицировать пациентов с фенотипом низкой экскреции, вызванным конкретными патогенными вариантами (напр., p.M405V, p.V400M, p.R227P). Высокая частота встречаемости известных патогенных вариантов в определенных этнических группах делает возможным проведение экономически эффективной молекулярной диагностики и консультирование.
Пренатальную диагностику осуществляют путем определения повышенных концентраций глутаровой кислоты в околоплодных водах, путем анализа активности фермента в амниоцитах/образцах ворсинок хориона и путем идентификации известных патогенных вариантов GCDH.
4. Лизинурическая непереносимость белка (семейная непереносимость белка). Это редкое АуР-заболевание вызвано нарушением транспорта катионных аминокислот лизина, орнитина и аргинина в кишечнике и почках. Дефицит транспортного белка (транспортер-1 аминокислоты Y+L) при этом заболевании вызывает мультисистемные проявления, которые первоначально начинаются с желудочно-кишечных симптомов. Транспортный дефект при этой патологии находится в базолатеральной (антилюминальной) мембране энтероцитов и эпителии почечных канальцев. Это объясняет невозможность проникновения катионных аминокислот через эти клетки, даже если они вводятся в форме дипептидов. Лизин в форме дипептида проникает через люминальную мембрану энтероцитов, но гидролизуется с образованием свободных молекул лизина в цитоплазме. Свободный лизин, который не может пройти сквозь базолатеральную мембрану клеток, диффундирует обратно в просвет.
Вскоре после рождения могут появляться отказ от еды, тошнота, отвращение к белку, рвота и легкая диарея, которые приводят к задержке физического развития, истощению и гипотонии. Дети, находящиеся на грудном вскармливании, обычно не имеют симптомов вплоть до отлучения от груди, возможно, из-за низкого содержания белка в грудном молоке. Эпизоды гипераммониемии могут возникать после приема пищи с высоким содержанием белка. Обычными симптомами у пациентов недиагностированной патологией являются гепатоспленомегалия легкой/средней степени, остеопороз, редкие ломкие волосы, тонкие конечности с умеренным ожирением туловища и задержка роста. Развитие нейрокогнитивных функций обычно соответствует норме, но у некоторых пациентов наблюдалась умеренная умственная отсталость.
У пациентов с этой патологией часто возникает прогрессирующий интерстициальный пневмонит с приступами тяжелого обострения. Это состояние обычно прогрессирует до тяжелого альвеолярного протеиноза. К клиническим проявлениям относятся прогрессирующая одышка при ФН, утомляемость, кашель, ослабленное дыхание и хрипы на вдохе. У пациентов старшего возраста может развиться цианоз. У некоторых пациентов диагноз не был установлен вплоть до появления легочных симптомов. У 65% пациентов наблюдались рентгенологические признаки легочного фиброза, при этом клинические проявления легочного поражения отсутствовали.
Поражение почек первоначально проявляется протеинурией, гематурией и повышением уровня креатинина в сыворотке крови. Заболевание может прогрессировать до почечной недостаточности терминальной стадии. Также может развиться поражение почечных канальцев с лабораторными признаками почечного синдрома Фанкони (Fanconi). Биопсия почек выявляет патологические признаки, соответствующие гломерулонефриту и тубулоинтерстициальному нефриту. Могут наблюдаться гематологические признаки анемии, лейкопении, тромбоцитопении и повышенного содержания ферритина. Кроме того, сообщалось о состоянии, напоминающем гемофагоцитарный лимфогистиоцитоз/синдром активации макрофагов. Частыми признаками лизинурической непереносимости белка являются иммунологические отклонения (нарушение функции лимфоцитов, аномалии Ig, гипокомплементемия) и острый панкреатит.
К лабораторным признакам относятся гипераммониемия и повышенная концентрация оротовой кислоты в моче, которые возникают после приема пищи с высоким содержанием белка. Концентрации лизина, аргинина и орнитина в плазме крови обычно слегка понижены, но при этом значительно повышается содержание этих аминокислот (особенно лизина) в моче. Патогенез гипераммониемии, вероятно, обусловлен истощением промежуточных продуктов цикла мочевины, вызванным слабой абсорбцией и повышенной потерей орнитина и аргинина в почках. Концентрации аланина, глутамина, серина, глицина и пролина в плазме обычно повышены. Частыми признаками являются анемия, повышенное содержание ферритина, ЛДГ, тироксинсвязывающего глобулина в сыворотке крови, гиперхолестеринемия и гипертриглицеридемия. Это заболевание следует отличать от гипераммониемии, вызванной дефектами цикла мочевины, особенно у гетерозиготных женщин с дефицитом орнитинтранскарбамилазы при отсутствии повышенной экскреции лизина, орнитина и аргинина с мочой.
Лечение низкобелковой диетой, обеспечивающей рекомендуемую суточную норму белка, с приемом цитруллина (50-100 мг/кг в сутки) может привести к биохим. и клиническим улучшениям. Эпизоды гипераммониемии следует лечить незамедлительно. Лизин (10-30 мг/кг в сутки) в малых и частых дозах позволяет улучшить его показатели в плазме. При появлении у пациентов боли в животе и диареи дозу лизина следует уменьшить. У некоторых пациентов лечение острых легочных осложнений высокими дозами преднизона оказалось эффективным. Бронхолегочный лаваж следует назначать пациентам с альвеолярным протеинозом. Заболевание характерно для Финляндии и Японии, где частота встречаемости составляет 1:60 000 и 1:57 000 живорождений соответственно.
Ген непереносимости лизинурического белка (SLC7A7) расположен в хромосоме 14q11.2. Беременность женщин с этой патологией осложнялась анемией, тромбоцитопенией, токсемией и кровотечением, но дети рождались здоровыми.