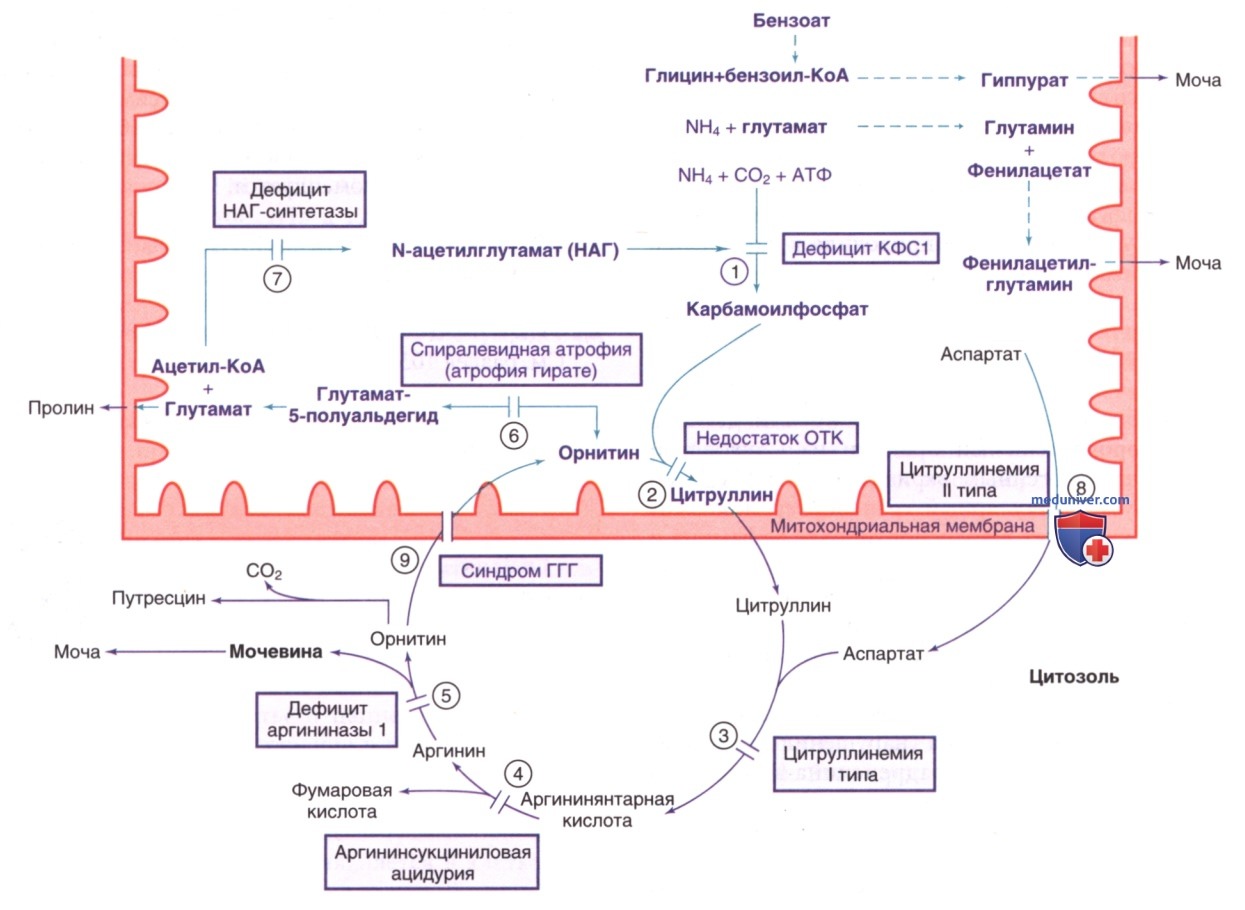
Катаболизм аминокислот приводит к образованию свободного аммиака, высокие концентрации которого токсичны для ЦНС. Млекопитающие нейтрализуют аммиак до мочевины посредством серии реакций, известных как цикл мочевины (см. рис. 12), в котором участвуют пять ферментов: карбамоилфосфатсинтетаза-1, орнитинтранскарбамилаза, аргининосукцинатсинтетаза, аргининосукцинатлиаза и аргиназа-1.
Рисунок 12. Цикл мочевины: пути экскреции аммиака и метаболизма орнитина. Реакции, протекающие в митохондриях, показаны фиолетовым цветом. Реакции, показанные пунктирными стрелками, представляют собой альтернативные пути экскреции аммиака. Ферменты: 1) карбамиолфосфатсинтетаза типа 1 (КФС1); 2) орнитинтранскарбамилаза (ОТК); 3) аргининосукцинатсинтетаза (АСС); 4) аргининосукцинатлиаза (АСЛ); 5) аргиназа 1 ; 6) орнитинаминотрансфераза; 7) N-ацетилглутамат (НАГ) синтетаза; 8) цитрин; 9) транспортер орнитина (0РНТ1). Синдром ГГГ — синдром гиперорнитинемии-гипераммониемии-гомоцитруллинемии
Шестой фермент, N-ацетилглутаматсинтетаза, катализирует синтез НАГ, который является обязательным активатором (эффектором) фермента карбамоилфосфатсинтетазы-1. Сообщалось об отдельных дефицитах этих ферментов. Согласно оценкам, частота встречаемости этой патологии составляет 1:35 000 живорожденных. Указанные нарушения являются наиболее частой генетической причиной гипераммониемии у младенцев.
а) Генетические причины гипераммониемии. Врожденные нарушения метаболизма, отличные от дефектов цикла мочевины, также могут приводить к гипераммониемии, иногда в тяжелой форме (табл. 4, 5). Механизмы гипераммониемии при некоторых из этих состояний разнообразны и включают накопление токсичных метаболитов (напр., органических кислот), нарушение транспорта промежуточных продуктов цикла мочевины (напр., синдром гиперорнитинемии-гиперам-мониемии-гомоцитруллинемии) и истощение промежуточных продуктов цикла мочевины (напр., лизинурическая непереносимость белка), что приводит к нарушению функции цикла мочевины.
б) Клинические проявления гипераммониемии. В неонатальном периоде симптомы и признаки обусловлены преимущественно дисфункцией ЦНС и схожи независимо от причины гипераммониемии. Новорожденный с данной патологией при рождении выглядит обычным, но после введения белкового прикорма появляются симптомы заболевания. Отказ от еды, рвота, тахипноэ и вялость могут быстро прогрессировать до глубокой комы. Часто появляются судороги. При физикальном обследовании отмечается снижение болевой чувствительности, а также гепатомегалия. Гипераммониемия может вызвать повышение ВЧД, которое может проявляться выбуханием родничка и расширением зрачков.
У младенцев и детей старшего возраста острая гипераммониемия проявляется рвотой и неврологическими нарушениями, такими как атаксия, спутанность сознания, раздражительность, повышенная возбудимость, агрессивность и психоз. Эти проявления могут чередоваться с периодами вялости и сонливости, способными прогрессировать до комы.
Если гипераммониемия вызвана дефектами ферментов цикла мочевины, то плановые лабораторные исследования не выявляют каких-либо специфических признаков. Концентрация АМК у таких пациентов обычно снижена. У некоторых пациентов первоначально может необъяснимо повышаться АЛТ и ACT в сыворотке крови и даже соответствовать критериям острой печеночной недостаточности. У младенцев с органическими ацидемиями гипераммониемия обычно сопровождается тяжелым ацидозом и кетонурией. У новорожденных с гипераммониемией часто ошибочно диагностируют сепсис; без верного диагноза эти дети могут погибнуть. Методы нейровизуализации позволяют выявить ОГМ.
При вскрытии можно обнаружить микровезикулярный стеатоз, холестаз легкой степени тяжести и фиброз печени. Т.о., нарушения цикла мочевины имеют неспецифические проявления, и поэтому необходимо измерять концентрацию аммиака в плазме крови любого младенца с тяжелым сепсисом, необъяснимой дисфункцией печени, рецидивирующей рвотой и прогрессирующей энцефалопатией.
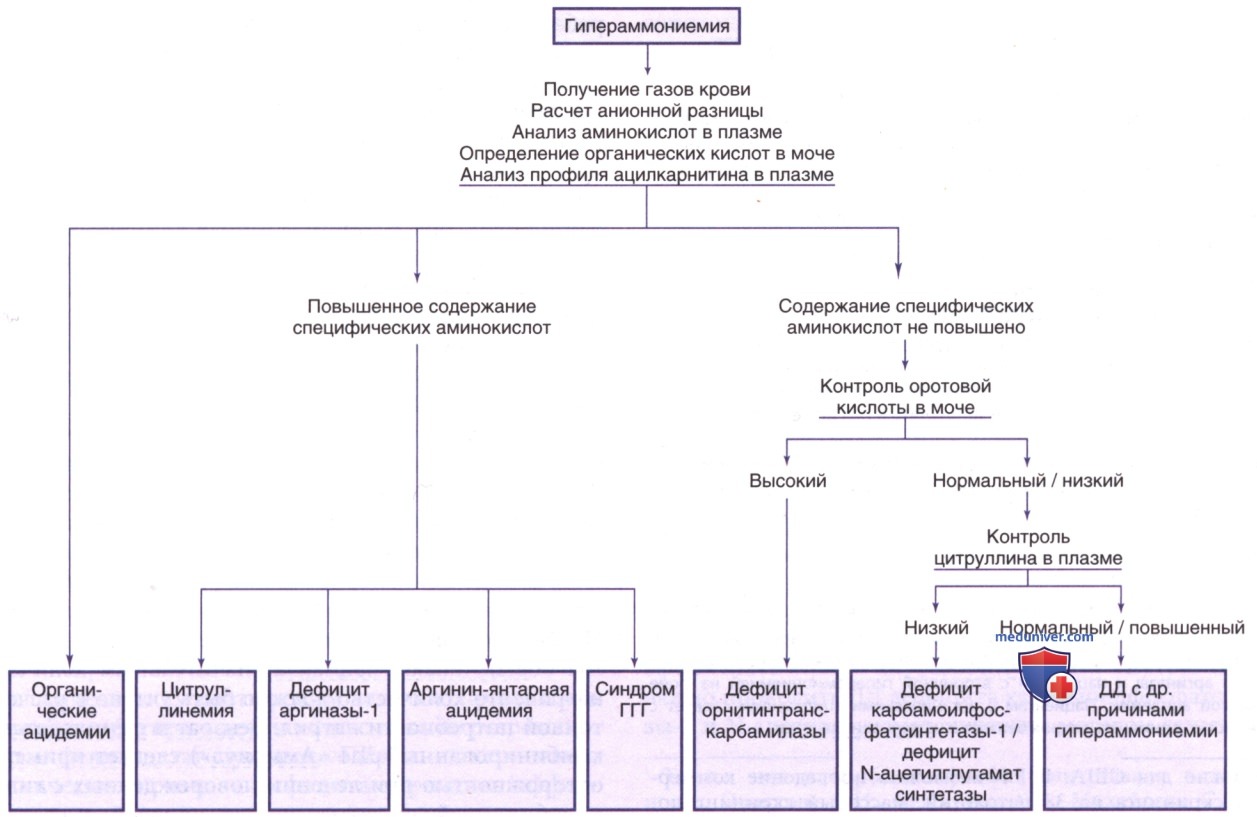
в) Диагностика. Главный ДК — гипераммониемия. Каждая клиническая лаборатория устанавливает свои нормы показателей аммиака в крови. Нормы данного показателя у новорожденных выше, чем у детей старшего возраста/взрослых. У здоровых доношенных детей могут наблюдаться концентрации до 100 мкмоль/л. У новорожденного с патологией уровень аммиака в крови обычно >150 мкмоль/л. На рис. 13 показан подход к ДД гипераммониемии у новорожденного. Помочь в постановке диагноза может тщательный анализ отклонений от нормы отдельных аминокислот в плазме.
Рисунок 13. Клинический подход к новорожденному с симптомами гипераммониемии. Синдром ГГГ — синдром гиперорнитинемии-гипераммониемии-гомоцитруллинемии
У пациентов с дефицитом карбамоилфосфатсинтетазы-1, орнитинтранскарбамилазы и N-ацетилглутаматсинтетазы частыми ДК являются повышение концентрации глутамина и аланина в плазме при одновременном снижении цитруллина и аргинина. Указанные нарушения нельзя отличить друг от друга только по содержанию аминокислот в плазме. Дефицит орнитинтранскарбамилазы можно дифференцировать от дефицита карбамоилфосфатсинтетазы-1 по значительному увеличению оротовой кислоты в моче.
С целью ДД дефицита карбамоилфосфатсинтетазы-1 и дефицита N-ацетилглутаматсинтетазы проводят определение соответствующих ферментов/молекулярно-генетическое исследование. Указывать на дефицит N-ацетилглутаматсинтетазы может клиническое улучшение после приема внутрь карбамилглутамата. У пациентов с дефицитом аргининосукцинатсинтетазы, аргининосукцинатлиазы и аргиназы 1 наблюдается заметное повышение в плазме концентраций цитруллина, аргининянтарной кислоты и аргинина соответственно. Сочетание гипераммониемии и выраженной гиперцитруллинемии/ аргинин-янтарной ацидурии фактически является патогномоничным признаком данных заболеваний.
Дети с нарушениями цикла мочевины часто самостоятельно придерживаются диеты с низким содержанием белка и высоким содержанием углеводов, особенно дети с поздним началом заболевания/женщины с клинической симптоматикой и частичным дефицитом орнитинтранскарбамилазы.
Массовый скрининг новорожденных позволяет выявить пациентов с дефицитом аргининосукцинатсинтетазы, аргининосукцинатлиазы и аргиназы-1*.
P.S. * Актуально для США. В РФ возможно проведение коммерческого скрининга на 38 патологий, массовый скрининг новорожденных предполагает пять заболеваний (гипотиреоз, адреногенитальный синдром, фенилкетонурия, муковисцидоз, галактоземия).
г) Лечение острой гипераммониемии. Клиническая картина в основном зависит от тяжести и продолжительности гипераммониемии. Тяжелые неврологические осложнения могут возникнуть у новорожденных со значительно повышенным уровнем аммиака в крови (>300 мкмоль/л) >12 ч. Поэтому острую гипераммониемию следует лечить быстро и интенсивно. Целью терапии является снижение концентрации аммиака. Это достигается двумя способами: 1) выведение аммиака из организма в форме, отличной от мочевины, и 2) минимизация эндогенного распада белка и способствование эндогенному синтезу белка за счет обеспечения достаточного количества калорий и незаменимых аминокислот (табл. 5).
Необходимо в/в введение жидкости, электролитов, декстрозы («Глюкозы») (10-15%) и липидов (1-2 г/кг в сутки) в сочетании с минимальным количеством белка (0,25 г/кг в сутки), предпочтительно с незаменимыми аминокислотами. Как только станет заметно улучшение, следует начинать энтеральное кормление смесью с низким содержанием белка (0,5-1,0 г/кг в сутки) через назогастральный зонд.
Поскольку почки плохо нейтрализуют аммиак, его выведение из организма должно ускоряться за счет образования соединений с высоким почечным клиренсом. Важным достижением в лечении гипераммониемии стало внедрение терапии ацилирования с использованием экзогенной органической кислоты, которая эндогенно ацилируется заменимыми аминокислотами с образованием нетоксичного соединения с высоким почечным клиренсом. Основными органическими кислотами, используемыми для этой цели, являются натриевые соли бензойной и фенилуксусной кислот. Бензоат с эндогенным глицином в печени образует гиппурат (см. рис. 12). Каждый моль бензоата удаляет 1 моль аммиака в виде глицина.
Фенилацетат соединяется с глутамином с образованием фенилацетилглутамина, который легко выводится с мочой. Один моль фенилацетата выводит из организма 2 моля аммиака в виде глутамина (см. рис. 12). Основным ЛП для приема внутрь является натрия фенилбутират, метаболизирующийся до фенилацетата. Для в/в применения в продаже имеется комбинированный состав бензоата и фенилацетата («Аммонуд»).
Еще одним важным средством терапии является в/в ведение аргинина, эффективное у всех пациентов (за исключением пациентов с дефицитом аргиназы). Введение аргинина поставляет орнитин для цикла мочевины (см. рис. 12). У пациентов с цитруллинемией 1 моль аргинина реагирует с 1 молем аммиака (в виде карбамоилфосфата) с образованием цитруллина. У пациентов с аргинин-янтарной ацидурией 2 моля аммиака (в виде карбамоилфосфата и аспартата) реагируют с аргинином с образованием аргинин-янтарной кислоты. Цитруллин и аргининосукцинат менее токсичны, чем аммиак, и легче выводятся почками.
Пациентам с дефицитом карбамоил-фосфатсинтетазы-1/орнитинтранскарбамилазы требуется введение аргинина, поскольку эта аминокислота не вырабатывается в достаточных количествах для эндогенного синтеза белка.
Для энтеральной терапии пациенты с дефицитом орнитинтранскарбамилазы используют БАД цитруллина (200 мг/кг в сутки), при этом 1 моль цитруллина реагирует с 1 молем аммиака (через аспарагиновую кислоту) с образованием аргинина. Аргинин/цитруллин противопоказаны пациентам с дефицитом аргиназы — редким состоянием, при котором обычная клиническая картина представляет собой не гипераммониемию, а спастическую диплегию. Терапия аргинином оказывается неэффективной, если гипераммониемия развивается на фоне органической ацидемии. У новорожденного с дебютом гипераммониемии следует применять аргинин до установления диагноза (см. табл. 5).
Для достижения максимального терапевтического эффекта можно вводить совместно бензоат, фенилацетат и аргинин. После первой дозы данных ЛП следует непрерывно проводить инфузию до выхода пациента из острого состояния. И бензоат, и фенилацетат обычно выпускаются в виде концентрированных р-ров, и для в/в введения их необходимо разбавить надлежащим образом (1-2% р-р). Рекомендуемые терапевтические дозы обоих ЛП обеспечивают пациента значительным количеством натрия; это количество должно быть учтено в расчете суточной потребности натрия.
Бензоат и фенилацетат (или комбинированный ЛП «Аммонул») следует применять с осторожностью при лечении новорожденных с гипербилирубинемией, поскольку может произойти вытеснение билирубина из альбумина; тем не менее среди новорожденных с гипераммониемией, получавших такое лечение, нет зарегистрированных случаев керниктеруса. Младенцам из группы риска рекомендуется снизить билирубин до безопасного уровня, учитывая в/в введение бензоата/фенилацетата.
Если исходный уровень аммиака <500 мкмоль/л и вышеуказанные методы лечения не дают заметных изменений уровня аммиака в крови в течение 4-6 ч, следует проводить гемодиализ. Для пациентов с уровнем аммиака >500 мкмоль/л стартовым методом удаления аммиака является экстракорпоральная детоксикация. Заменное переливание крови мало влияет на снижение общего уровня аммиака в организме. Его следует применять только в случае невозможности проведения диализа в кратчайшие сроки/если пациентом является новорожденный ребенок с гипербилирубинемией (см. выше).
Гемодиализ резко снижает уровень аммиака в крови в течение нескольких часов, но, если он недоступен/технически невозможен, в качестве альтернативы можно использовать перитонеальный диализ. Если гипераммониемия вызвана органической ацидемией, а гемодиализ недоступен, можно использовать перитонеальный диализ для выведения патогенной органической кислоты и аммиака.
Прием неомицина внутрь ограничивает рост кишечных бактерий, продуцирующих аммиак. Однако у пациентов (напр., у новорожденных с этой патологией), которым срочно требуется снижение уровня аммиака, этот метод применяется ограниченно. Прием лактулозы внутрь подкисляет среду в просвете кишечника, т.о. уменьшая диффузию аммиака через эпителий кишечника. Этот ЛП имеет ограниченное применение у новорожденных с высоким риском ацидемии и обезвоживания.
В качестве дополнительной терапии у новорожденных с метаболической энцефалопатией, напр., вызванной гипераммониемией, было предложено использовать охлаждение. Для оценки эффективности данного подхода проводятся клинические исследования. Улучшение неврологического статуса после нормализации уровня аммиака требует времени. Иногда это занимает несколько дней.
- Долгосрочная терапия гипераммониемии. Как только у младенца купируются неврологические признаки, следует проводить терапию основной причины гипераммониемии. В целом всем пациентам, независимо от ферментативного дефекта, необходимо ограничить белок в пределах рекомендуемой нормы потребления с поправкой на возраст. Детям с нарушениями цикла мочевины для поддержания уровня аммиака в крови в пределах нормы (указанные дозы приведены для пациентов с МТ <20 кг) эффективно регулярное введение бензоата натрия (250 мг/кг в сутки), натрия фенилбутирата (250-500 мг/кг в сутки) и аргинина (200-400 мг/кг в сутки) и цитруллина (пациентам с дефицитом орнитинтранскарбамилазы 200-400 мг/кг в сутки).
Пациентам с аргининемией противопоказаны аргинин и цитруллин. Пациентам при проблемах с приемом натрия .фенилбутирата можно провести пробу с глицеринфенилбутиратом. Это соединение скрывает неприятный запах натрия фенилбутирата и позволяет пациенту соблюдать режим лечения. Применение глицеринфенилбутирата детям <2 мес пока не разрешено. Бензоат и фенилацетат снижают содержание карнитина, но клинические признаки дефицита карнитина/эффективность приема карнитина пока только предстоит установить. Данные ЛП назначались беременным женщинам и очевидного тератогенного эффекта не наблюдалось. Однако опыт применения пока еще ограничен, поэтому следует проявлять соответствующую осторожность при назначении.
Необходимо внимательно следить за показателями физического развития, особенно окружности головы и показателей питания (исследование крови на альбумин, преальбумин, pH, электролиты, аминокислоты, цинк, селен). Наиболее эффективное долгосрочное лечение таких пациентов достигается при совместной работе опытных специалистов (педиатра, диетолога, детского невролога, генетика). У нескольких пациентов с разл. типами дефектов цикла мочевины наблюдались поражения кожи, напоминающие энтеропатический акродерматит, предположительно из-за дефицита незаменимых аминокислот, вызванного чрезмерным ограничением белков в рационе. Следует избегать катаболических состояний (инфекции, голодание), способных вызвать гипераммониемию. При их появлении необходимо начинать интенсивное лечение.
Всем детям с нарушениями цикла мочевины важно исключить назначение вальпроевой кислоты, поскольку этот ЛП может повышать уровень аммиака в крови даже у некоторых ЗЛ. Введение вальпроата пациентам с дефицитом карбамоилфосфатсинтетазы-1, орнитинтранскарбамилазы и аргининосукцинатсинтетазы может спровоцировать обострение гипераммониемии.
д) Дефициты карбмамоилфосфатсинтетазы-1 и N-ацетилглутаматсинтазы. Дефициты карбамоилфосфатсинтетазы-11 и N-ацетил-глутаматсинтазы вызывают сходные клинические и биохим. проявления (см. рис. 12, 13). Степень выраженности симптомов и возраст дебюта сильно различаются. Почти полный дефицит ферментов проявляется в первые несколько дней/часов жизни в виде признаков и симптомов гипераммониемии (отказ от еды, рвота, гипотония, судороги, кома). Нередко повышается ВЧД. Поздние формы (до 40 лет) могут проявляться у внешне ЗЛ в виде острого приступа гипераммониемии (гипотония, головная боль, судороги, психоз). Такие эпизоды могут приводить к развитию комы и даже смерти (ранее бессимптомная 26-летняя женщина умерла от гипераммониемии во время родов).
Диагноз часто путают с мигренью. Также наблюдались промежуточные формы, сопровождающиеся умственной отсталостью и хронической субклинической гипераммониемией с периодическими приступами острой гипераммониемии.
К лабораторным признакам относится гипераммониемия. Исследование состава аминокислот в плазме крови обычно показывает отчетливое повышение глутамина и аланина при относительно низких уровнях цитруллина и аргинина. Эти изменения не относятся к диагностическим, т.к. гипераммониемия возникает по разным причинам. В моче обычно понижено содержание/полностью отсутствует оротовая кислота (см. рис. 13).
Лечение кризов гипераммониемии и длительная терапия этого заболевания описаны ранее (см. табл. 5). У пациентов с дефицитом N-ацетилглутаматсинтетазы эффективен прием карбамилглутамата внутрь. Поэтому важно проводить ДД дефицита карбамоилфосфатсинтетазы-1 и N-ацетилглутаматсинтетазы методом генетического секвенирования. Дефицит N-ацетилглутаматсинтетазы в Северной Америке встречается редко.
Дефицит карбамоилфосфатсинтетазы-1 и N-ацетилглутаматсинтетазы наследуется по АуР-типу; фермент карбамоилфосфатсинтетаза-1 представлен в печени и кишечнике. Ген (CPS1) расположен в хромосоме 2q34. Частота встречаемости этого заболевания составляет 1:1 300 000. Ген N-ацетилглутаматсинтетазы (NAGS) расположен в хромосоме 17q21.31. Ни одно из этих заболеваний не выявляется при массовом скрининге новорожденных.
е) Орнитинтранскарбамилазная недостаточность. При этом Х-сцепленном наследственном заболевании гемизиготные мальчики имеют более тяжелые проявления, чем гетерозиготные девочки (см. рис. 12, 13). У гетерозиготных девочек может наблюдаться легкая форма, но большинство (75%) являются бессимптомными носителями, хотя исследования указывают на незначительные неврологические нарушения даже у девочек без гипераммониемии в анамнезе. Наиболее частой формой всех нарушений цикла мочевины является дефицит орнитинтранскарбамилазы, который составляет 40% случаев нарушений цикла мочевины.
Клинические проявления у новорожденных мальчиков обычно в виде тяжелой гипераммониемии, возникающей в первые несколько дней жизни. Легкие формы, как у некоторых гетерозиготных девочек, обычно имеют эпизодические проявления, которые могут возникнуть в любом возрасте (обычно >1 года). Эпизоды гипераммониемии в виде рвоты и неврологических нарушений (напр. атаксии, спутанности сознания, возбуждения, агрессивности, манифестного психоза) чередуются с периодами хорошего самочувствия. Указанные эпизоды обычно возникают после приема пищи с высоким содержанием белка/в результате катаболического состояния, напр., инфекции. Один из таких приступов может прогрессировать до гипераммониемической комы, ОГМ и смерти.
Умственное развитие может соответствовать норме. Тем не менее часто встречается умственная отсталость легкой/средней степени. У выживших развивается ЖКБ; механизм неясен.
Основным лабораторным признаком во время криза является гипераммониемия, сопровождающаяся заметным повышением концентрации глутамина и аланина в плазме, в то время как содержание цитруллина и аргинина остается низким. Концентрация мочевины в крови обычно низкая. Данное заболевание дифференцируют с дефицитом карбамоилфосфатсинтетазы-1 по значимому увеличению экскреции оротовой кислоты с мочой (см. рис. 13). Оротат может выпадать в осадок в моче в виде розового песка/камней. При легкой форме лабораторные отклонения могут нормализоваться в межприступный период. Следует проводить ДД такой формы со всеми эпизодическими заболеваниями детского возраста.
В частности, у пациентов с лизинурической непереносимостью белка могут наблюдаться некоторые признаки дефицита орнитинтранскарбамилазы, но первую из указанных патологий можно отличить по повышенной экскреции лизина, орнитина и аргинина с мочой и повышенной концентрации цитруллина в крови.
Диагноз легче всего подтвердить генетическим исследованием. Однако у 20% пациентов с данной патологией наблюдается нормальная последовательность ДНК, возможно, из-за того, что дефект гена включает варианты мутаций с участием интронов/промоторной области. Варианты числа копий патогенных аллелей можно определить методом хромосомного микроматричного анализа (молекулярного кариотипирования), и в случае «+» результата следует рассмотреть вопрос о возможности делеции сцепленного гена. При «-» результате молекулярной диагностики м.б. показана биопсия печени. Пренатальную диагностику осуществляют методом ДНК-анализа в ам-ниоцитах/образцах ворсинок хориона.
Повышение экскреции оротидина с мочой после нагрузочного теста с аллопуринолом позволяет выявлять носителей патологии среди женщин. У бессимптомных женщин-носителей может присутствовать минимальная мозговая дисфункция. Следует подчеркнуть, что имеет значение детальный наследственный анамнез. У родственниц пробанда по материнской линии часто наблюдаются мигрень/отвращение к белковой пище. Более того, тщательное изучение семейного анамнеза позволяет выявить закономерность необъяснимой смерти новорожденных мальчиков по материнской линии.
Ранее было описано лечение острых приступов гипераммониемии и долгосрочная терапия этого заболевания. Для энтерального применения пациентам с дефицитом орнитинтранскарбамилазы вместо аргинина назначают цитруллин. Эффективным способом лечения пациентов с тяжелым дефицитом орнитинтранскарбамилазы является трансплантация печени.
Ген орнитинтранскарбамилазы картирован в Х-хромосоме (Хр21.1). Выявлено множество мутаций, вызывающих заболевание (>300). Частота встречаемости дефицита орнитинтранскарбамилазы составляет 1:56 000-77 000 живорожденных. Генотип и, как следствие, степень дефицита фермента в большинстве случаев определяют выраженность фенотипа. Предполагается, что матери младенцев с данной патологией будут носителями мутантного гена, если мутация не возникает de novo. Было выявлено, что мать двух мальчиков с данной патологией обладала нормальным генотипом, что позволяет предположить гонадный мозаицизм в некоторых семьях. Дефицит орнитинтранскарбамилазы не выявляется при массовом скрининге новорожденных.
ж) Цитруллинемия. Выявлены две разные формы цитруллинемии — как клинически, так и генетически. Классическая форма (I тип) вызвана дефицитом фермента аргининосукци-натсинтетазы. Цитруллинемии II типа вызвана дефицитом митохондриального транспортного белка цитрина (см. рис. 12, 13).
1. Цитруллинемия I типа (дефицит аргининосукцинатсинтетазы, классическая цитруллинемия). Данное заболевание вызвано дефицитом аргининосукцинатсинтетазы (см. рис. 12). Существуют разл. клинические проявления в зависимости от степени дефицита фермента. Выявлены две основные формы. Наиболее часто встречается тяжелая/неонатальная форма, которая проявляется в первые несколько дней жизни признаками и симптомами гипераммониемии (см. ранее). При под-острой/легкой форме клинические проявления (такие как задержка физического и психомоторного развития, частая рвота и сухие, ломкие волосы) появляются постепенно >1 года. Диагноз можно заподозрить при появлении острой гипераммониемии, вызванной интеркуррентным катаболическим состоянием.
Лабораторные данные аналогичны таковым у пациентов с дефицитом орнитинтранскарбамилазы, за исключением концентрации цитруллина в плазме, которая в этом случае значительно повышена (в 50-100 раз выше нормы) (см. рис. 13). Экскреция оротовой кислоты с мочой умеренно повышена; кроме того, в результате осаждения оротатов может наблюдаться кристаллурия. Диагноз подтверждают ДНК-исследованием/реже — анализом активности ферментов в культуре фибробластов. Пренатальную диагностику проводят методом ферментативного анализа культуры амниотических клеток/анализа ДНК клеток, полученных при биопсии ворсинок хориона.
Лечение острых приступов гипераммониемии и длительная терапия описаны ранее (см. табл. 5). Концентрация цитруллина в плазме все время остается повышенной и в дальнейшем может увеличиваться после введения аргинина. Пациенты могут иметь хорошее самочувствие при соблюдении низкобелковой диеты в сочетании с терапией бензоатом натрия, фенилбутиратом и аргинином. Когнитивные расстройства легкой/средней степени тяжести являются частым осложнением даже у пациентов с надлежащим лечением.
Цитруллинемия наследуется по АуР-типу. Ген (ASS1) расположен в хромосоме 9q34.11. У большинства пациентов наблюдаются сложные гетерозиготные мутации по двум разл. аллелям. Частота встречаемости заболевания составляет 1:250 000 живорожденных. Недавнее введение неонатального скрининга на дефекты цикла мочевины показало, что некоторые пациенты с данной патологией кажутся здоровыми даже при обычном рационе. Необходимо длительное наблюдение для уверенности, что у таких пациентов отсутствуют неврологические осложнения.
2. Дефицит цитрина (цитруллинемия II типа). Цитрин (транспортный белок аспартата-глутамата) является митохондриальным транспортером, кодируемым геном (SLC25A13), расположенным в хромосоме 7q21.3. Одной из функций данного белка является транспорт аспартата из митохондрий в цитоплазму и восполнение цитозольного пула аспартата, необходимого для превращения цитруллина в аргинин-янтарную кислоту (см. рис. 12). Если аспартат недоступен для цитоплазматического компонента цикла мочевины, мочевина не сможет образовываться с нормальной скоростью, и цитруллин будет накапливаться. У таких пациентов снижена активность аргининосукцинатсинтетазы в печени, но патогенный вариант в гене ASS1 не был обнаружен.
Предполагается, что дефицит цитрина препятствует трансляции матричной РНК для фермента аргининосукцинатсинтетазы в печени.
Заболевание первоначально было зарегистрировано в Японии, но впоследствие обнаружены и пациенты неяпонского происхождения. Описаны две клинические формы дефицита цитрина.
- Неонатальный внутрипеченочный холестаз (цитруллинемия II типа, неонатальная форма). Клинические и лабораторные проявления, которые обычно начинаются <1 года, включают холестатическую желтуху с прямой (конъюгированной) гипербилирубинемией легкой/средней степени, выраженную гипопротеинемию, нарушения свертывания (увеличенное ПВ и АЧТВ), а также повышение уровня у-глутамилтрансферазы и ЩФ; трансаминазы в печени обычно в норме. Содержание аммиака и цитруллина в плазме крови обычно в норме, хотя зарегистрированы случаи умеренного повышения. В сыворотке крови может наблюдаться повышение концентрации метионина, тирозина, аланина и треонина; а также повышенные уровни галактозы, хотя ферменты метаболизма галактозы остаются в норме. Этиология гипергалактоземии неизвестна. Наблюдается заметное повышение уровня АФП в сыворотке крови.
Данные признаки напоминают проявления тирозинемии I типа, но, в отличие от последней, экскреция сукцинилацетона с мочой не повышена. При гист. исследовании биоптатов печени наблюдается жировая инфильтрация, холестаз с расширенными протоками и фиброз средней степени выраженности. Заболевание обычно проходит самостоятельно, и большинство младенцев спонтанно выздоравливают к 1 году при поддерживающем и симптоматическом лечении. В нескольких случаях развилась печеночная недостаточность, требующая трансплантации печени. Хотя данное заболевание чаще всего наблюдается в Японии, заподозрить патологию следует в случае любого необъяснимого неонатального гепатита с холестазом. Данные о долгосрочном прогнозе и патогенезе заболевания ограничены. Отмечается, что неонатальный в/пече-ночный холестаз переходит во взрослую форму после нескольких лет кажущейся ремиссии.
- Цитруллинемия II типа, взрослая форма (цитруллинемия у взрослых; цитруллинемия II типа, легкая форма). Цитруллинемия II типа дебютирует у ранее казавшихся ЗЛ достаточно остро и проявляется нейропсихиатрическими симптомами, такими как дезориентация, делирий, бред, аберрантное поведение, тремор и манифестный психоз. Наблюдается умеренная гипераммониемия и гиперцитруллинемия. Возраст начала заболевания обычно 20-40 лет (от 11 до >100 лет). Пациенты, выздоровевшие после первого эпизода, могут страдать от рецидивирующих приступов. Основными осложнениями у выживших являются панкреатит, гиперлипидемия и гепатома.
Лечение чаще всего не предотвращало последующие приступы. Восстановить цитозольный пул аспартата и стимулировать выработку мочевины помогает диета, обогащенная белками и липидами. Однако предполагается, что введение большого количества глюкозы может оказывать «-» влияние, т.к. для гликолитического пути важен транспортер цитрина. Хотя трансплантация печени, по-видимому, эффективна для профилактики последующих эпизодов гипераммониемии, в качестве стартовой терапии рекомендуется прием внутрь аргинина, пирувата и среднецепочечных триглицеридов для облегчения эпизодов гипераммониемии и нормального развития.
В японских и неяпонских семьях с данной патологией было обнаружено несколько мутаций гена, вызывающих заболевание. Хотя гомозиготность в Японии встречается относительно часто (1:20 000 человек), частота встречаемости клинических проявлений заболевания составляет лишь 1:100 000-230 000 человек. Это означает, что у значительного числа гомозиготных носителей симптомы отсутствуют.
з) Дефицит аргининосукцинатлиазы (аргини-янтарная ацидурия). Выраженность клинических и биохим. проявлений значительно варьирует (см. рис. 12, 13). При тяжелой форме дефицита аргининосукцинатлиазы признаки и симптомы тяжелой гипераммониемии (см. ранее) развиваются в первые несколько дней жизни, при отсутствии лечения часто приводят к летальному исходу. Клиническое течение дефицита аргининосукцинатлиазы у пациентов, переживших дебют острого приступа (криза), может характеризоваться умственной отсталостью, задержкой физического развития, АГ, болезнями пищевого тракта, печеночным фиброзом и гепатомегалией. Часто у таких пациентов при отсутствии лечения наблюдаются сухие и ломкие волосы (узловатый трихорексис*).
При катаболическом статусе могут возникать острые приступы тяжелой гипераммониемии.
P.S. * Узловатый трихорексис — дефект волосяного стержня, характеризующийся утолщением/слабыми местами, из-за которых волосы легко ломаются. Эта группа состояний способствует выпадению, отсутствию роста и повреждению волос.
К лабораторным признакам относятся гипераммониемия, умеренное повышение ферментов печени, неспецифическое повышение уровней глутамина и аланина в плазме, умеренное повышение уровня цитруллина в плазме (меньше, чем при цитруллинемии) и заметное увеличение концентрации аргинин-янтарной кислоты в плазме, моче и СМЖ. Концентрация в последней обычно выше, чем в плазме. Фермент обычно присутствует в эритроцитах, печени и культуре фибробластов. Пренатальная диагностика основана на измерении активности фермента в культуре амниотических клеток/на выявлении мутации в гене ASL. В околоплодных водах плода с данной патологией повышен уровень аргинин-янтарной кислоты.
Лечение острых приступов гипераммониемии и длительная терапия данного заболевания описаны ранее в этой статье выше. Распространенными осложнениями являются умственная отсталость, стойкая гепатомегалия с умеренным повышением уровня ферментов печени и повышенная кровоточивость вследствие нарушения синтеза факторов свертывания крови. Данный дефицит наследуется по АуР-типу. Частота встречаемости составляет 1:220 000 живорожденных. Ген (ASL) расположен в хромосоме 7q11.21. Массовый скрининг новорожденных позволяет диагностировать патологию на ранней стадии**.
P.S. ** Актуально для США.
и) Дефицит аргиназы-1 (гипераргининемия). Данный дефект наследуется по АуР-типу (см. рис. 12, 13). У людей есть две генетически разл. аргиназы. Одна из них цитозольная (ARG1) и экспрессируется в печени и эритроцитах, а вторая (ARG2) присутствует в митохондриях почек и ГМ. Ген фермента ARG1 (дефицит которого наблюдается у пациентов с недостаточностью аргиназы-1) расположен в хромосоме 6q23.2. Роль митохондриального фермента не до конца известна; его активность возрастает у пациентов с аргинине-мией, но не оказывает протективного эффекта.
Клинические проявления этого редкого дистального нарушения цикла мочевины несколько отличаются от таковых при др. дефектах ферментов цикла мочевины, хотя были описаны случаи острой неонатальной формы с плохо поддающимися терапии судорогами, ОГМ и летальным исходом. Дебют дефицита аргиназы-1 часто бывает скрытым; заболевание может оставаться бессимптомным в первые несколько месяцев/лет. Прогрессирующая спастическая диплегия, ножницеобразные ноги, хореоатетоидные движения, утрата ранее приобретенных навыков и задержка физического развития у ранее здорового ребенка могут указывать на дегенеративное заболевание ЦНС. Некоторые дети годами получали лечение ДЦП, прежде чем у них был подтвержден дефицит аргиназы 1.
Прогрессирует умственная отсталость, часто проявляются судороги, но эпизоды тяжелой гипераммониемии возникают не так часто, как при более проксимальных дефектах цикла мочевины. Может наблюдаться гепатомегалия.
К лабораторным ДК относят заметное повышение уровня аргинина в плазме и СМЖ (см. рис. 13). В моче м.б. повышено содержание оротовой кислоты. Важнейшим этапом диагностики аргининемии является определение состава аминокислот в плазме. В моче увеличивается содержание гуанидиносоединений (а-кетогуанидиновалериановой кислоты и а-кетоаргининовой кислоты). Для подтверждения диагноза определяют активность аргиназы в эритроцитах/выяв-ляют мутацию гена.
Лечение заключается в назначении низкобелковой диеты в рамках рекомендованной суточной нормы потребления. Состав диеты и суточное потребление белка контролируют частым динамическим определением аминокислот в сыворотке крови. Для контроля гипераммониемии и снижения уровня аргинина в плазме также эффективны натрия бензоат/натрия фенилбутират. Умственная отсталость является распространенным осложнением данного заболевания. У одного пациента, несмотря на тщательный контроль, к 9 годам развился СД-1. Трансплантация печени показывает многообещающие результаты, но опыт долгосрочных последствий ограничен. Массовый скрининг новорожденных позволяет диагностировать патологию на ранней стадии*.
P.S. * Актуально для США.
к) Транзиторная гипераммониемия новорожденного. Концентрация аммиака в крови доношенных новорожденных может достигать 100 мкмоль/л/быть в 2-3 раза выше, чем у детей старшего возраста/взрослых. Через несколько недель жизни норма уровня аммиака в крови приближается к таковым значениям у взрослых (см. рис. 13).
У некоторых новорожденных наблюдается тяжелая форма транзиторной гипераммониемии. Большинство младенцев с данной патологией — это недоношенные с РДС легкой степени. Гипераммониемическая кома может развиться в течение 2-3 сут и при отсутствии лечения приводит к летальному исходу. Лабораторно наблюдаются выраженная гипераммониемия (уровень аммиака в плазме достигает 4000 мкмоль/л) и умеренное повышение концентраций глутамина и аланина в плазме. Содержание в плазме промежуточных в цикле мочевины аминокислот обычно остается в норме, за исключением цитруллина, который м.б. умеренно повышенным. Причина нарушения неизвестна. Активность ферментов цикла мочевины в норме. Лечение гипераммониемии следует начинать незамедлительно и проводить интенсивно. Обычно удается достичь выздоровления без каких-либо осложнений, а эпизоды гипераммониемии не рецидивируют даже при обычном белковом рационе.
л) Нарушение метаболизма орнитина. Орнитин — ключевой промежуточный продукт цикла мочевины, не входит в состав природных белков. Скорее всего, он образуется в цитозоле из аргинина и транспортируется в митохондрии, где становится субстратом для реакции, катализируемой орнитинтранскарбамилазой, с образованием цитруллина. Избыток орнитина катаболизируется двумя ферментами: орнитинаминотрансферазой (митохондриальным ферментом, превращающим орнитин в предшественник пролина) и орнитиндекарбоксилазой (цитозольный фермент, превращающий орнитин в путресцин) (см. рис. 12). Гиперорнитинемия характеризуется двумя генетическими нарушениями: атрофией сетчатки и синдромом гипераммониемии-гиперорнитинемии-гомоцитруллинемии.
1. Гиратная* атрофия сетчатки и сосудистой оболочки. Это редкое АуР-заболевание вызвано дефицитом орнитинаминотрансферазы (см. рис. 12). Приблизительно 30% всех зарегистрированных случаев приходится на Финляндию. У некоторых пациентов в первый месяц жизни может наблюдаться гипераммониемия. Клинические проявления, определяющие фенотип дефицита орнитинаминотрансферазы, включают ночную слепоту, миопию, потерю периферического зрения и заднекапсулярную катаракту. Данные нарушения дебютируют в 5-10 лет и прогрессируют до полной слепоты к 40 годам. Атрофические изменения сетчатки напоминают извилины ГМ. При такой патологии интеллект обычно не страдает. Помимо характерного 10-20-кратного увеличения концентрации орнитина в плазме (400-1400 мкмоль/л), концентрации глутамата, глутамина, лизина, креатина и креатинина в плазме м.б. умеренно снижены.
Некоторые пациенты частично отвечают на терапию высокими дозами пиридоксина. Диета с ограничением аргинина в сочетании с добавлением лизина, пролина и креатина позволяет снижать концентрацию орнитина в плазме и способствует некоторому клиническому улучшению. Ген орнитинаминотрансферазы (ОАТ) расположен в хромосоме 10q26.13. В разных семьях были выявлены разл. мутации гена (не <60).
2. Синдром гипераммониемии-гиперорнитинемии-гомоцитруллинемии. При этом редком АуР-заболевании нарушен транспорт орнитина из цитозоля в митохондрии, что приводит к накоплению орнитина в цитозоле и истощению этой аминокислоты в митохондриях. Первое вызывает гиперорнитинемию, а второе — нарушение цикла мочевины и гипераммониемию (см. рис. 12). Гомоцитруллин предположительно образуется в результате реакции митохондриального карбамоилфосфата с лизином, который может выступать субстратом для реакции орнитинтранскарбамилазы при дефиците орнитина. Клиническая симптоматика гипераммониемии может развиться вскоре после рождения/в зрелом возрасте. Острые эпизоды гипераммониемии проявляются отказом от еды, рвотой и летаргией; в младенчестве может приводить к коме.
Если заболевание остается недиагностированным, то могут прогрессировать неврологические симптомы, такие как гипотония мышц нижних конечностей, усиление глубоких сухожильных рефлексов, спастические и клонические судороги и задержка психомоторного развития разл. степени выраженности. У пациентов с синдромом гипераммониемиигиперорнитинемии-гомоцитруллинемии не наблюдалось никаких симптомов заболеваний глаз. Лабораторное исследование выявляет значимое повышение концентраций орнитина и гомоцитруллина в плазме в сочетании с гипераммониемией (см. рис. 13). Острые эпизоды гипераммониемии требуют незамедлительного лечения (см. ранее). При гипераммониемии эффективна низкобелковая диета. Прием аргинина (или цитруллина) внутрь у некоторых пациентов приводил к клиническому улучшению. Ген синдрома гипераммониемии-гиперорнитинемиигомоцитруллинемии (SLC25A15) расположен в хромосоме 13q14.11.
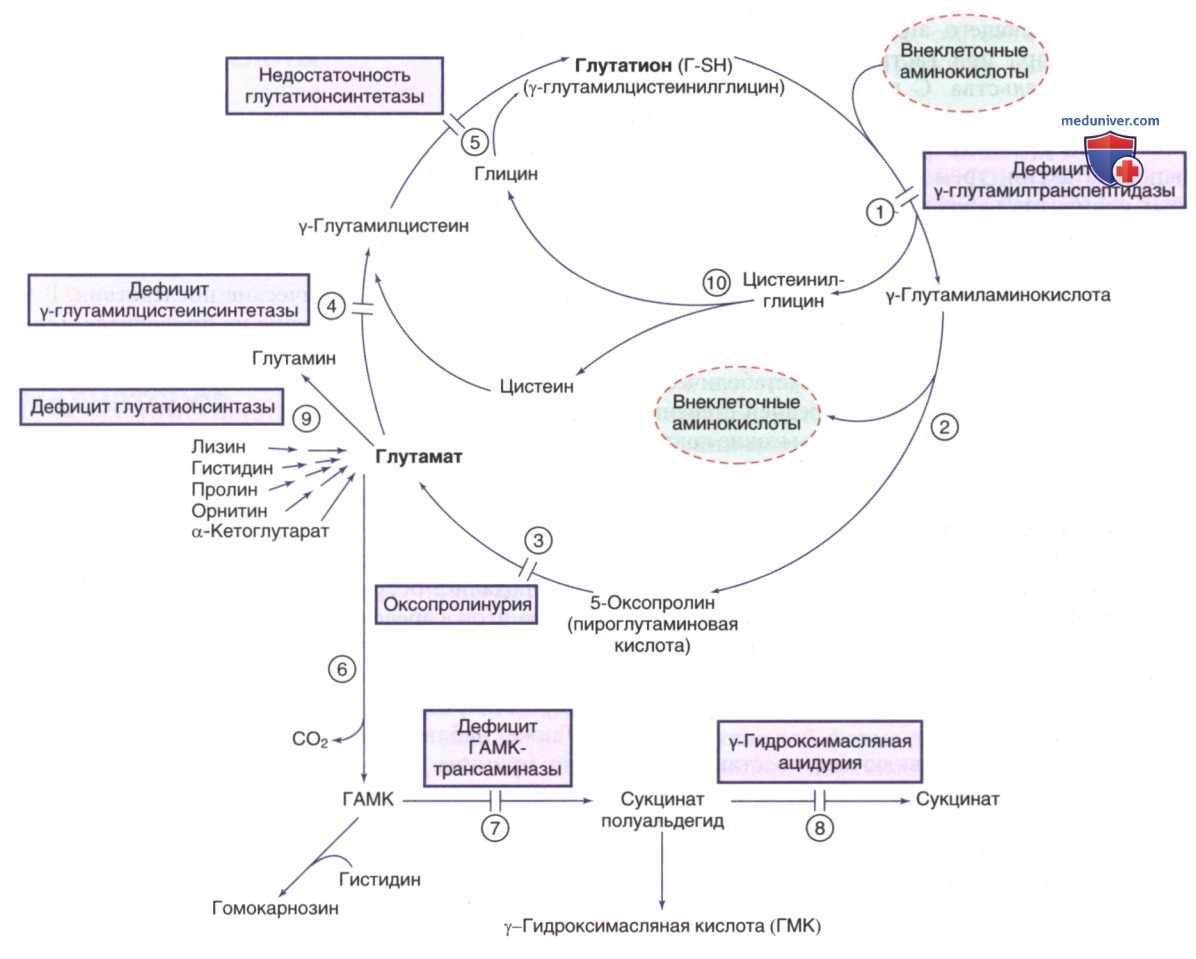
м) Врожденный дефицит глутамина. Глутамин синтезируется эндогенно из глутамата и аммиака широко представленным в организме ферментом глутаминсинтетазой (см. рис. 11). Известно, что глутамин задействован в нескольких важных функциях, включая детоксикацию аммиака. Дефицит данного фермента, приводящий к дефициту глутамина, был зарегистрирован у трех младенцев из трех неродственных семей. У всех детей с такой патологией при рождении наблюдалась полиорганная патология, в т.ч. тяжелые ВПР ГМ (аномальные вращения, гипомиелинизация), лицевые аномалии (широкая переносица, низкое расположение ушных раковин), гипотония и судороги. Двое новорожденных умерли от СПОН (ДН и СН). Один ребенок дожил до 3 лет, но страдал тяжелой задержкой психомоторного развития. Глутамин отсутствовал в плазме, моче и СМЖ, при этом содержание глутаминовой кислоты в плазме было в норме.
Рисунок 11. γ-Глутамиловый цикл и связанные с ним пути. Отмечены нарушения синтеза и распада глутатиона (Г-SH). Ферменты: 1) γ-глутамилтранспептидаза (ГГТ); 2) у-глутамилциклотрансфераза; 3) 5-оксопролиназа; 4) у-глутамилцистеинсинтетаза; 5) глутатионсинтетаза; 6) декарбоксилаза глутаминовой кислоты; 7) трансаминаза гамма-аминомасляной кислоты (ГАМК); 8) сукцинат-полуальдегиддегидрогеназа; 9) глутаминсинтетаза; 10) дипептидаза
Генетические мутации этого фермента подчеркивают важнейшую роль глутамина в эмбриогенезе, особенно для нормального развития ГМ. Заболевание наследуется по АуР-типу; ген глутаминсинтетазы (GLUL) расположен в хромосоме 1q25.3.