Митохондриальное β-окисление жирных кислот является важным путем обеспечения организма энергией. Этот процесс приобретает особенно большое значение при длительном голодании и в периоды пониженного потребления калорий, связанные с заболеваниями ЖКТ или с повышенным расходом энергии при лихорадочных заболеваниях. В этих состояниях организм переходит от преимущественного использования углеводов в качестве основного вида топлива к преимущественному использованию жиров. Кроме того, жирные кислоты имеют большое значение в качестве топлива для работающих скелетных мышц и являются предпочтительным субстратом нормального метаболизма сердца.
В этих тканях жирные кислоты полностью окисляются до диоксида углерода и воды. Конечными продуктами окисления жирных кислот в печени являются кетоновые тела — β-гидроксибутират и ацетоацетат. Они не могут окисляться в печени, но экспортируются в периферические ткани и служат в них важным источником топлива, особенно в ГМ, где кетоновые тела могут частично заменять глюкозу в периоды голодания.
Выявлены генетические дефекты, которые затрагивают почти все стадии окисления жирных кислот. Почти для всех характерен рецессивный путь наследования (табл. 1).
Клинические проявления выявляются, как правило, со стороны тканей, в которых активно протекают процессы β-окисления: печени, скелетных мышц и сердца. Чаще всего такие заболевания проявляются острым коматозным состоянием, печеночной энцефалопатией и гипогликемией на фоне длительного голодания, это связано с нарушением кетогенеза в печени. Часто наблюдается хроническая кардиомиопатия и мышечная слабость или рабдомиолиз, спровоцированный ФН. Вне периодов голодания или повышенной потребности в энергии дефекты окисления жирных кислот могут не проявляться.
Можно ошибочно диагностировать внезапно возникшие симптомы заболевания, такие как синдром Рейе или, в случае летального исхода, синдром внезапной детской смерти. Нарушения окисления жирных кислот легко пропустить, поскольку до проведения специальных метаболических исследований единственным ключом к диагнозу м.б. обнаружение несоответствующих низких концентраций кетонов плазмы или мочи у младенца с гипогликемией. Генетические дефекты утилизации кетоновых тел также можно не заметить, поскольку при гипогликемии натощак кетонемия вполне ожидаема. В некоторых ситуациях клинические проявления в большей степени обусловлены токсическим воздействием метаболитов жирных кислот, а не дефицитом энергии.
Такие ситуации возникают при определенных нарушениях окисления длинноцепочечных жирных кислот [при дефиците длинноцепочечной 3-гидроксиацил-дегидрогеназы (LCHAD), карнитинпальмитоилтрансферазы-IA (CPT-IA) или митохондриального трифункционального белка (МТР, известного также как TFP)]. При таких нарушениях наличие пораженного гомозиготного плода увеличивает риск жизнеугрожающего заболевания у беременной гетерозиготной матери. В результате развивается острый жировой гепатоз беременных (ОЖГБ) или преэклампсия с HELLP-синдромом (гемолиз, повышенные уровни ферментов печени, низкий уровень тромбоцитов).
Механизм указанных акушерских осложнений, вероятно, связан с накоплением токсических промежуточных метаболитов. При тяжелой недостаточности электрон-переносящего флавопротеина (ETF), ETF-дегидрогеназы (ETF-DH) и карнитинпальмитоилтрансферазы II (СРТ-П) описаны мальформации ГМ и почек, которые м.б. связаны с в/утробной токсичностью метаболитов жирных кислот или с ролью вышеуказанных ферментов в процессе эмбрионального развития. При дефиците LCHAD и МТР описаны прогрессирующая дистрофия сетчатки, периферическая невропатия и хронические прогрессирующие заболевания печени. При большинстве таких нарушений программы скрининга новорожденных с использованием тандемной масс-спектрометрии позволяют обнаружить характерные ацилкарнитиновые профили плазмы и установить диагноз на ранней стадии, до появления симптомов.
Использование скрининговых программ показало, что все нарушения окисления жирных кислот в совокупности являются самыми распространенными врожденными нарушениями метаболизма, по крайней мере в популяциях, в основном у людей европеоидной расы.
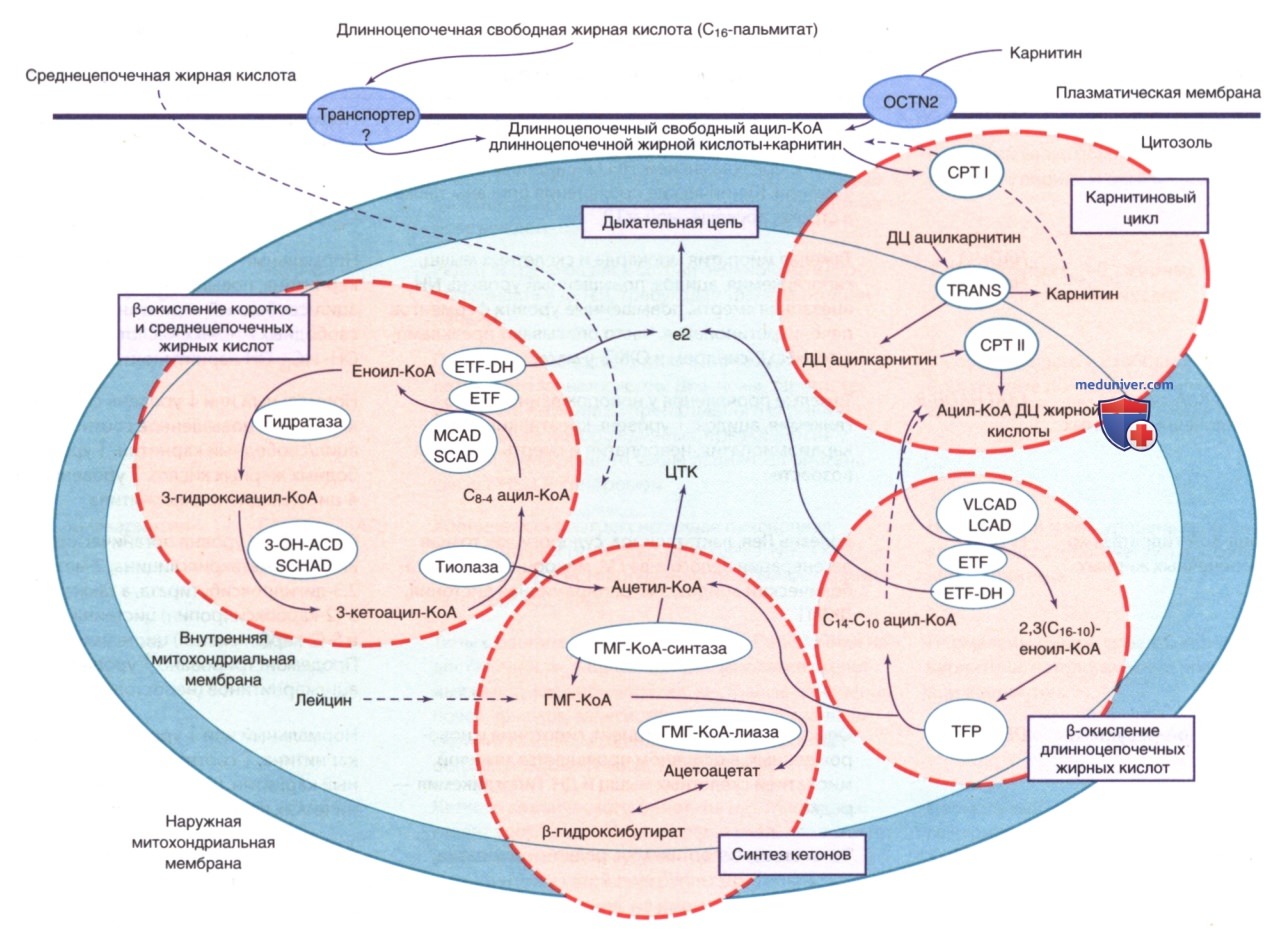
На рис. 1 и 2 кратко описаны этапы окисления типичной длинноцепочечной жирной кислоты. В карнитиновом цикле длинноцепочечные жирные кислоты транспортируются через барьер, образованный внутренней мембраной митохондрий, в виде эфиров ацил-карнитина. (Среднецепочечные жирные кислоты, которые часто назначают младенцам с нарушением питания в виде ЛП среднецепочечных триглицеридов, могут напрямую вступать в цикл митохондриального β-окисления, минуя карнитиновый цикл). В митохондриях в результате четырехэтапного цикла β-окисления коэнзим А (КоА)-активированные жирные кислоты превращаются в единицы ацетил-КоА. На каждом этапе β-окисления требуется два или три разных специфических изофермента для единиц ацил-КоА с жирными кислотами разной длины.
Рисунок 1. Митохондриальное окисление жирных кислот. Карнитин входит в клетку с помощью транспортера органических катионов/карнитина (OCTN2). Пальмитат, типичная 16-углеродная длинноцепочечная жирная кислота, переносится через клеточную мембрану и может активироваться с образованием ацилкоэнзима А (КоА) длинноцепочечных (LC) жирных кислот. После этого ацил-КоА длинноцепочечных жирных кислот вступает в карнитиновый цикл, в котором переэтерифицируется под действием пальмитоилтрансферазы I (СРТ-I), переносится через внутреннюю митохондриальную мембрану с помощью карнитин/ацилкарнитинтранслоказы (TRANS), а затем под действием карнитинпальмитоилтрансферазы II (СРТ-II) снова превращается в ацил-КоА длинноцепочечных жирных кислот, чтобы подвергнуться р-окислению. Под действием ацил-КоА-дегидрогеназы жирных кислот с очень длинной цепью (VLCAD/LCAD) образуется (С16-С10) 2,3-еноил-КоА. Митохондриальный трифункциональный протеин (МТР) выполняет функции еноил-КоА-гидратазы (гидратазы), 3-ОН-гидроксиацил-КоА-дегидрогеназы (3-ОН-АКД) и р-кетотиолазы (тиолазы). При участии МТР образуются ацетил-КоА, восстановленная форма флавинадениндинуклеотида (FADH) и восстановленная форма никотинамиддинуклеотида (NADH). Средне- и короткоцепочечные жирные кислоты (С8-4) могут входить в митохондриальный матрикс независимо от карнитинового цикла. Для этого требуется участие ацил-КоА-дегидрогеназы среднецепочечных жирных кислот (MCAD), ацил-КоА-дегидрогеназы короткоцепочечных жирных кислот (SCAD) и гидроксиацил-КоА-дегидрогеназы короткоцепочечных жирных кислот (SCHAD). После этого ацетил-КоА может вступать в цикл Кребса (ЦТК). При участии электронпереносящего флавопротеина (ETF) и дегидрогеназы электронпереносящего флавопротеина (ETF-DH) электроны переносятся с никотинамидадениндинуклеотида (НАДН) на дыхательную цепь. НАДН входит в цепь переноса электронов через комплекс I. В печени ацетил-КоА может превращаться в гидроксиметилглутарил-(ГМГ)-КоА при помощи β-гидрокси-β-метилглутарил-КоА-синтазы (ГМГ-КоА-синтазы), а затем, при участии β-гидрокси-β-метилглутарил-КоА-лиазы (ГМГ-КоА-лиазы), в ацетоацетат, относящийся к кетоновым телам
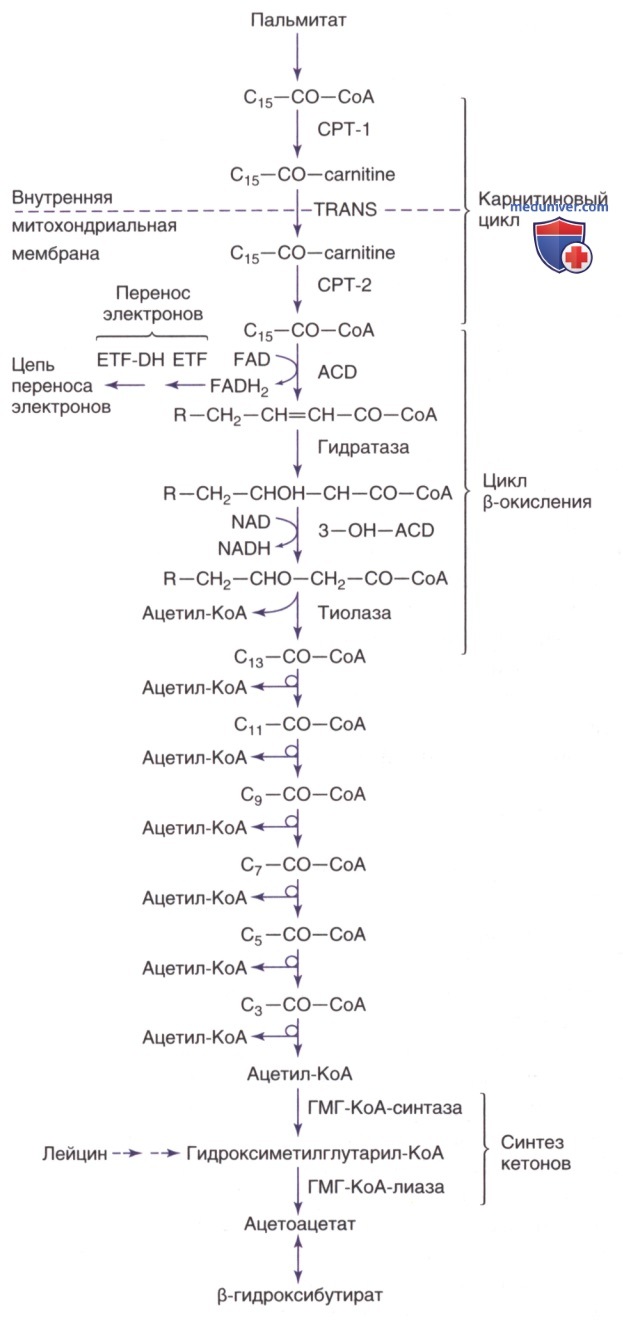
Рисунок 2. Путь митохондриального окисления пальмитата —типичной 16-углеродной длинноцепочечной жирной кислоты. Ферментные этапы включают карнитинпальмитоилтрансферазу (СРТ) 1-го и 2-го типов, карнитин/ацилкарнитинтранслоказу (TRANS), электронпереносящий флавопротеин (ETF), ETF-дегидрогеназу (ETF-DH), ацил-КоА-дегидрогеназу (ACD), еноил-КоА-гидратазу (гидратазу), 3-гидроксиацил-КоА-дегидрогеназу (3-0H-ACD), β-кетотиолазу (тиолазу), β-гидрокси-β-метилглутарил-КоА (ГМГ-КоА)-синтазу и лиазу; NAD — ни-котинамидадениндинуклеотид; NADH — никотинамидадениндинуклеотид (восстановленный); FAD — флавинадениндинуклеотид FADH2 — восстановленная форма флавинадениндинуклеотида
Электроны, образовавшиеся на первом этапе β-окисления (ацил-КоА-дегидрогеназа), перемещаются на цепь переноса электронов на уровне коэнзима Q посредством пути переноса электронов для участия в синтезе аденозинтрифосфата. Электроны, образовавшиеся на третьем этапе (3-гидроксиацил-КоА-дегидрогеназа), перемещаются на цепь переноса электронов на уровне комплекса 1. Основная часть ацетил-КоА, образовавшегося в результате β-окисления жирных кислот в печени, проходит через путь кетогенеза, в результате которого образуются β-гидроксибутират и ацетоацетат, а в мышцах и сердце жирные кислоты полностью окисляются до СО2 и воды.
а) Дефекты в цикле β-окисления:
1. Дефицит ацил-КоА-дегидрогеназы среднецепочечных жирных кислот. Дефицит ацил-КоА-дегидрогеназы среднецепочечных жирных кислот (MCAD) является наиболее распространенным нарушением окисления жирных кислот. Для этого нарушения продемонстрирован выраженный «эффект основателя». У большинства пациентов имеются северозападноевропейские корни, а большинство из пациентов с такими корнями являются гомозиготными по единственной общей миссенс-мутации в гене MCAD — замене нуклеотида А на G в позиции 985 в комплементарной ДНК (кДНК) (c.985A>G). Вследствие этой мутации вместо лизина на остатке 329 синтезируется глутаминовая кислота (р.К329Е).
- Клинические проявления. У пациентов с ранее не установленным диагнозом заболевание, как правило, проявляется в период с первых 3 мес жизни до 5 лет острыми эпизодами, провоцируемыми длительным голоданием (>12-16 ч). Для заболевания характерны такие объективные и субъективные симптомы, как рвота и сонливость, быстро прогрессирующие до судорог и острой сердечно-легочной недостаточности. Может быть внезапная детская смерть. Печень м.б. слегка увеличена и содержать жировые включения. В первые несколько месяцев жизни приступы редкие. Предположительно это связано с большей частотой кормлений в более раннем возрасте. В более старшем возрасте риск возникновения приступов увеличивается в связи с прекращением ночных кормлений и голоданием в период интеркуррентных заболеваний детского возраста.
Зарегистрированы случаи, когда заболевание проявлялось в первые дни жизни гипогликемией новорожденных. Такие случаи отмечали у детей, которые по недосмотру оставались голодными, или у детей, находящихся на грудном вскармливании, при позднем прикладывании к груди. Зарегистрированы редкие случаи диагностирования дефицита MCAD у ранее ЗЛ. Это означает, что даже при отсутствии симптомов в период младенчества сохраняется риск метаболической декомпенсации при достаточно длительном голодании. Некоторое количество пациентов могут оставаться бессимптомными. Ок. 25% пациентов с дефицитом MCAD умирают или получают серьезное повреждение ГМ в результате первого эпизода, еще до проведения рутинного скрининга новорожденных. В настоящее время диагноз у большинства пациентов устанавливают в период новорожденности по результатам скринингового исследования мазка крови на ацилкарнитины.
Это позволяет начать терапию на ранних сроках заболевания и предотвратить развитие многих тяжелых субъективных и объективных симптомов. По данным некоторых сообщений, клинические проявления у новорожденных с дефицитом MCAD возникают остро, до получения результатов неонатального скрининга. У новорожденных, находящихся на исключительно грудном вскармливании, риск выше, поскольку в первые дни они получают мало калорий.
- Лабораторные признаки. Острые эпизоды, как правило, сопровождаются гипогликемией. Отмечаются несоответствующе низкие концентрации кетонов плазмы и мочи (гипокетотическая гипогликемия). В связи с гипокетонемией метаболический ацидоз, которого можно было бы ожидать у многих детей с гипогликемией, выражен слабо или вообще отсутствует. Результаты печеночных проб отклоняются от нормы — повышены уровни ферментов печени (АЛТ, ACT), повышены уровни аммиака крови, увеличены показатели длительности ПТВ и АЧТВ. При исследовании биоптатов печени, взятых в период острого заболевания, обнаруживают микро- или макровезикулярный стеатоз, обусловленный накоплением триглицеридов.
При определении профилей органических кислот мочи методом газовой хроматографии/масс-спектрометрии в периоды голодания или острого заболевания обнаруживают несоответствующе низкие концентрации кетонов и повышенные уровни среднецепочечных дикарбоновых кислот (адипиновой, пробковой и себациновой кислот), которые получаются в результате омега-окисления накопленных среднецепочечных жирных кислот в микросомах и пероксисомах. Концентрации общего карнитина в плазме крови и тканях снижены до 25-50% от нормы, а фракция общего эстерифицированного карнитина повышена. Такой характер вторичного дефицита карнитина наблюдают при большинстве дефектов окисления жирных кислот. Он является отражением конкуренции между повышенными уровнями ацилкарнитина и свободного карнитина за транспорт через плазматическую мембрану почечных канальцев. К значимым исключениям из этого правила относятся дефицит мембранного транспортера карнитина, CPT-IA или β-гидрокси-β-метилглутарил-КоА (ГМГ-КоА)-синтазы, который не проявляется вторичной карнитиновой недостаточностью.
При дефиците MCAD диагностическое значение имеют следующие паттерны метаболитов; повышенные уровни ацилкарнитинов плазмы С6:0, С8:0 С10:0 и С10:1 и повышенные уровни ацилглицинов мочи, в т.ч. гексаноилглицина, субе-рилглицина и 3-фенилпропионилглицина. Неонатальный скрининг, который проводят почти всем детям, родившимся в США, позволяет обнаружить дефицит MCAD до появления симптомов по отклонению от нормы уровней ацилкарнитинов в пятне крови на фильтровальной бумаге. Диагноз можно подтвердить путем выявления распространенной мутации A985G или путем секвенирования гена MCAD. У детей, заболевание которых было выявлено в результате неонатального скрининга, обнаружили еще один распространенный вариант — Т199С. Интересно, что на данный момент этот аллель не определяли у пациентов, у которых дефицит MCAD сопровождался симптомами. Возможно, он является результатом более легкой мутации.
- Лечение. Острое заболевание требует немедленного начала терапии — в/в введения р-ров, содержащих 10% декстрозу («Глюкозу»), с целью коррекции или предотвращения гипогликемии и скорейшего подавления липолиза. Вне обострения терапия заключается в предотвращении голодания. Как правило, для этого требуется лишь скорректировать диету т.о., чтобы периоды ночного голодания не превышали 10-12 ч.
Эффективность ограничения потребляемых жиров или лечения ЛП карнитина сомнительна. Необходимость активных терапевтических вмешательств при варианте Т199С до сих пор не установлена.
- Прогноз. Почти у 25% пациентов с неустановленным диагнозом летальный исход может наступить во время первого приступа заболевания. В анамнезе таких пациентов нередко имеются данные о смерти братьев или сестер, предположительно связанной с недиагностированным дефицитом MCAD. У некоторых пациентов возможно постоянное повреждение ГМ во время приступов тяжелой гипогликемии. У тех выживших, у которых отсутствуют повреждения ГМ, прогноз благоприятный, поскольку для дефицита MCAD не характерны когнитивные нарушения и кардиомиопатия. С возрастом непереносимость голода становится менее выраженной, а риск возникновения приступов заболевания снижается. Поскольку у 35% пациентов с дефицитом MCAD заболевание никогда не проявлялось клинически, важно обследовать их братьев и сестер, чтобы выявить членов семьи, у которых оно протекает бессимптомно.
2. Дефицит ацил-КоА-дегидрогеназы жирных кислот с очень длинной цепью. Дефицит ацил-КоА-дегидрогеназы жирных кислот с очень длинной цепью (VLCAD) является вторым наиболее часто диагностируемым нарушением окисления жирных кислот. Изначально, до того, как узнали о существовании VLCAD, связанной с внутренней мембраной митохондрий, это заболевание называли дефицитом VLCAD. У всех пациентов, которым ранее установили диагноз дефицита VLCAD, имелись дефекты гена VLCAD. У пациентов с дефицитом VLCAD отсутствует способность к окислению физиол. длинноцепочечных жирных кислот. У них, как правило, более тяжелое поражение, чем у пациентов с дефицитом MCAD, у которых окислительные процессы нарушены в меньшей степени. Дефицит VLCAD проявляется в более раннем грудном возрасте и сопровождается более длительными симптомами, которые связаны с мышечной слабостью или эпизодами болей в мышцах и рабдомиолиза.
Во время острых приступов, провоцируемых голодом, может развиваться кардиомиопатия. Может отмечаться гипертрофия или дилатация ЛЖ, а также низкая сократительная способность по данным ЭхоКГ. У некоторых пациентов возникает внезапная смерть, однако состояние большинства переживших первый эпизод улучшается, нормализуется функция сердца. По остальным физикальным признакам и данным рутинных лабораторных исследований, в т.ч. по вторичному дефициту карнитина, заболевание сходно с MCAD. Профиль органических кислот мочи представлен некетотической дикарбоновой ацидурией с повышенными уровнями С6-12-дикарбоновых кислот. Диагноз можно предположить при отклонении ацилкарнитинового (С14:0, 14:1, 14:2) профиля от нормы по результатам исследования плазмы или мазка крови. Однако для установления конкретного диагноза требуется мутационный анализ гена VLCAD. Основной принцип лечения заключается в том, чтобы избегать голодания >10-12 ч. У некоторых пациентов эффективно непрерывное в/зондовое питание.
3. Дефицит ацил-КоА-дегидрогеназы короткоцепочечных жирных кислот. Описано небольшое число пациентов с разными фенотипами, у которых имелись две нулевые мутации в гене ацил-КоА-дегидрогеназы короткоцепочечных жирных кислот (SCAD). У большинства людей, отнесенных к имеющим дефицит SCAD, в действительности имеются полиморфные изменения ДНК в гене SCAD. Напр., к двум распространенным полиморфизмам относятся G185S и R147W. У 7% людей в популяции они представлены в гомозиготном виде. Некоторые исследователи утверждают, что эти варианты, возможно, являются факторами восприимчивости, и для их фенотипического проявления требуется вторая, пока неизвестная, генетическая мутация. Многие др. исследователи считают, что дефицит SCAD является безвредным биохим. расстройством. Данное расстройство с АуР-типом наследования проявляется неонатальной гипогликемией и может сопровождаться нормальными уровнями кетонов.
На диагноз указывают повышенные уровни бутилкарнитина (С4-карнитина) при исследовании отпечатков крови или плазмы крови у новорожденных, а также повышенные показатели экскреции этилмалоновой кислоты и бутирилглицина с мочой.
Перечисленные метаболические нарушения наиболее выражены у пациентов с нулевыми мутациями и присутствуют у некоторых гомозиготных пациентов по распространенным полиморфизмам.
До сих пор не установлено, существует ли необходимость в лечении дефицита SCAD. Было предложено обследовать бессимптомных лиц в течение длительного времени, чтобы определить, имеется ли у них реальное заболевание. У большинства людей с дефицитом SCAD симптомы заболевания могут не проявиться в течение всей жизни. Однако может существовать подгруппа людей с тяжелыми проявлениями — дисморфическими чертами лица, сложностями с питанием/отсутствием прибавки МТ, метаболическим ацидозом, кетотической гипогликемией, сонливостью, задержкой в развитии, судорогами, гипотонией, дистонией и миопатией.
4. Дефицит 3-гидроксиацил-КоА-дегидрогеназы длинноцепочечных жирных кислот/митохондриального трифункционального белка. Фермент LCHAD является частью МТР, который содержит еще два фермента, участвующих в этапах [3-окисления: 2,3-еноил-КоА-гидратазу длинноцепочечных жирных кислот и β-кетотиолазу длинноцепочечных жирных кислот. МТР представляет собой гетерооктамерный белок. Он состоит из четырех а- и четырех P-цепей, которые кодируются отдельными функционально сцепленными генами, делящими общую промоторную область. У некоторых пациентов заболевание затрагивает только функции LCHAD (дефицит LCHAD), тогда как у др. наблюдается дефицит всех трех функций МТР (дефицит МТР).
Клинические проявления включают приступы острой гипокетотической гипогликемии, сходные с приступами, которые наблюдаются при дефиците MCAD. У пациентов нередко отмечаются признаки более тяжелого заболевания — кардиомиопатии, мышечных судорог и мышечной слабости, нарушение функции печени (холестаз). Токсические эффекты метаболитов жирных кислот могут приводить к пигментной ретинопатии, которая может стать причиной слепоты, прогрессирующей печеночной недостаточности, периферической невропатии и рабдомиолиза. У гетерозиготных матерей с дефицитом LCHAD/МТР, вынашивающих гомозиготных детей, наблюдаются жизнеугрожающие акушерские осложнения (ОЖГБ, HELLP-синдром). М.б. внезапная детская смерть. На диагноз указывают повышенные уровни 3-гидроксиацилкарнитинов с длиной цепи С16-С18 по результатам исследования отпечатка крови или плазмы.
В профиле органических кислот мочи можно обнаружить повышенные уровни 3-гидроксикарбоновых кислот с длиной цепи С6-С10. Часто обнаруживают вторичный дефицит карнитина. Распространенную мутацию в гене, кодирующем α-субъединицу, наблюдают у >60% пациентов с дефицитом LCHAD. Эта мутация в организме плода особенно часто сопровождается акушерскими осложнениями, однако др. мутации в любой из субъединиц также связаны с заболеваниями у матери.
Лечение такое же, как при дефиците MCAD или VLCAD, т.е. следует избегать голодания. Некоторые исследователи высказали предположение об эффективности применения БАД, содержащих среднецепочечные триглицериды (чтобы обойти дефект окисления длинноцепочечных жирных кислот) и докозагексаеновую кислоту (для защиты от изменений сетчатки). Предпринимались попытки трансплантации печени у пациентов с тяжелой печеночной недостаточностью, однако это не приводило к уменьшению выраженности метаболических отклонений и не предотвращало миопатию или осложнения со стороны сетчатки.
5. Дефицит 3-гидроксиацил-КоА-дегидрогеназы короткоцепочечных жирных кислот. Зарегистрировано только 14 пациентов с подтвержденными мутациями 3-гидроксиацил-КоА-дегидрогеназы короткоцепочечных жирных кислот (SCHAD). В большинстве случаев рецессивные мутации в гене SCHAD проявлялись эпизодами гипокетотической гипогликемии, вызванной гиперинсулинизмом. Таким пациентам, в отличие от пациентов с др. формами нарушения окисления жирных кислот, для коррекции гиперинсулинизма и предотвращения повторных приступов гипогликемии требовалась специфическая терапия диазоксидом. У единственного пациента со сложными гетерозиготными мутациями в возрасте 10 лет развилась фульминантная печеночная недостаточность. Белок SCHAD обладает неферментной функцией, которая заключается в непосредственном взаимодействии с глутаматдегидрогеназой (ГДГ) и подавлении ее активности.
При отсутствии белка SCHAD активность ГДГ не подавляется и становится избыточной, что является признанной причиной гиперинсулинизма и, как правило, связано с активирующими мутациями гена GDH. Тяжелый дефицит белка SCHAD часто проявляется преимущественно белок-чувствительной гипогликемией, а не гипогликемией натощак. По-видимому, при наличии белка SCHAD ингибирование ГДГ сохраняется даже при отсутствии ферментной активности SCHAD. У таких пациентов могут проявляться более традиционные формы нарушений окисления жирных кислот. К специфическим метаболическим маркерам дефицита SCHAD относятся повышенные уровни С4-гидроксиацилкарнитина в плазме и 3-гидроксиглутаровой кислоты в моче. Исследование на дефицит SCHAD вошло в программу неонатального скрининга, однако чувствительность этого метода пока не установлена.
Пациентов с дефицитом SCHAD, у которых имеется гиперинсулинизм, лечат диазоксидом. Имеющегося опыта недостаточно для того, чтобы рекомендовать разные варианты лечения негиперинсулинемических форм дефицита SCHAD, однако предотвращение голодания представляется более целесообразным.
6. Дефицит 2,3-еноил-КоА-гидратазы короткоцепочечных жирных кислот. Это нарушение, возникающее в результате мутаций в гене ECHS1, выявлено недавно. Многих пациентов идентифицировали методом экзомного секвенирования. На данный момент в литературе описано ок. 20 случаев. Данное нарушение затрагивает общий путь метаболизма короткоцепочечных жирных кислот и валина. Клинические фенотипы более характерны для митохондриальных нарушений метаболизма пирувата, среди которых преобладает заболевание, напоминающее болезнь Лея и сопровождающееся выраженным, часто фатальным, лактатацидозом. На данный момент не найдено ни вариантов лечения, ни специфических биомаркеров. У нескольких пациентов обнаружено выделение повышенных количеств метакрилилглицина, высокореактивного и потенциально токсичного промежуточного метаболита, 2-метил-2,3-дигидроксибутирата, S-(2-карбоксипропил)-цистеина и S-(2-карбоксипропил)-цистеамина.
б) Нарушение карнитинового цикла:
1. Нарушение транспорта карнитина через плазматическую мембрану (первичный дефицит карнитина). Первичный дефицит карнитина — единственный генетический дефект, при котором дефицит карнитина является скорее причиной, чем следствием нарушенного окисления жирных кислот. Чаще всего этот дефект проявляется прогрессирующей кардиомиопатией, сопровождающейся или не сопровождающейся слабостью скелетных мышц, начинающейся в период с года до четырех лет. У меньшего числа пациентов до развития кардиомиопатии, сопровождающейся симптомами, в первый год жизни может наблюдаться гипокетотическая гипогликемия натощак. В основе кардиомиопатии лежат дефекты транспортера карнитина, зависимого от градиента натрия и расположенного в плазматической мембране клеток сердца, мышц и почек. Этот транспортер отвечает за поддержание в/клеточных концентраций карнитина на уровне, в 20-50 раз превышающем его концентрации в плазме крови, а также за сохранение карнитина в почках.
На диагноз дефекта транспортера карнитина указывают экстремально низкие уровни карнитина в плазме крови и мышцах (1-2% от нормы). У гетерозиготных родителей уровни карнитина в плазме крови составляют 50% от нормы. Кетогенез натощак может не отклоняться от нормы, поскольку не нарушен транспорт карнитина в печени, однако прекращение поступления карнитина с пищей может привести к нарушению кетогенеза натощак. При нарушении окисления жирных кислот в печени профиль органических кислот мочи натощак может демонстрировать гипокетотическую дикарбоновую ацидурию.
Др. значимых изменений профиля не наблюдается. Дефект транспорта карнитина может проявляться клинически выраженным снижением почечного порога карнитина. Кроме того, этот дефект можно обнаружить при исследовании захвата карнитина in vitro в культурах клеток фибробластов или лимфобластов. В основе этого заболевания лежат мутации в генах, кодирующих транспортер органических катионов/карнитина (OCTN2). Пероральная терапия фармакологическими дозами карнитина (100-200 мг/кг в сутки) высокоэффективна в отношении коррекции кардиомиопатии и мышечной слабости, а также нарушения кетогенеза натощак. На фоне лечения концентрации общего карнитина в мышцах остаются на уровне <5% от нормы.
2. Дефицит карнитин-пальмитоилтрансферазы IA. Дефицит изофермента CPT-I (CPT-IA) описан у нескольких десятков младенцев и детей. Клинические проявления включают индуцированную голоданием гипокетотическую гипогликемию, в редких случаях сопровождающуюся экстремальным отклонением результатов печеночных проб от нормы или почечным канальцевым ацидозом. Сердце и скелетные мышцы не вовлекаются в патологический процесс, поскольку мышечный изофермент не изменен. Профиль органических кислот мочи натощак в некоторых случаях демонстрирует гипокето-тическую С6-С12-дикарбоновую ацидурию, однако м.б. и нормальным.
При исследовании ацилкарнитина плазмы преобладает свободный карнитин и в небольшом количестве обнаруживается ацилированный карнитин. Этот факт используют для выявления дефицита CPT-IA в рамках неонатального скрининга методом тандемной масс-спектрометрии. Дефицит CPT-IA является единственной формой нарушения окисления жирных кислот, при которой уровень общего карнитина плазмы может быть повышен и нередко составляет 150-200% от нормы. Этот феномен объясняется отсутствием ингибирующего влияния длинноцепочечных ацилкарнитинов на транспортер карнитина в канальцах почек при дефиците CPT-IA. Эту ферментопатию можно продемонстрировать в культуре фибробластов или лимфобластов. Дефицит CPT-IA у плода, отмечавшийся в единственном доказанном случае, сопровождался ОЖГБ у матери.
Распространенный вариант гена CPT-IA (c.l436C>t, p.P479L) обнаружили у лиц инуитского происхождения, проживающих в США, Канаде и Гренландии. В популяции инуитов для этого варианта характерен повышенный риск синдрома внезапной детской смерти. Указанный вариант можно диагностировать с помощью неонатального скрининга. Активность фермента снижена на 80%, а регуляция малонил-КоА отсутствует. Не установлено, является ли CPT-IA (c.1436C>t, p.P479L) патологическим вариантом фермента или адаптацией к диете древних инуитов, содержащей большое количество жиров. Лечение при тяжелой форме дефицита CPT-IA, которую обнаружили у людей, не принадлежащих к популяции инуитов, сходно с лечением при дефиците MCAD и заключается в избегании ситуаций, в которых возникает кетогенез натощак. Есть ли необходимость в лечении при варианте, обнаруженном в популяции инуитов, не установлено.
3. Дефицит карнитин-ацилкарнитин-транслоказы. Дефект этого белка, переносящего длинноцепочечные ацилкарнитины через внутреннюю мембрану митохондрий, блокирует вхождение длинноцепочечных жирных кислот в митохондрии для участия в митохондриальном окислении. Клинический фенотип при этом расстройстве характеризуется тяжелым генерализованным нарушением окисления жирных кислот. В период новорожденности заболевание у большинства пациентов проявляется индуцированными голоданием гипогликемией, гипераммониемией и острой сердечно-легочной недостаточностью. У всех новорожденных, заболевание которых проявляется клинически, имеются признаки кардиомиопатии и мышечной слабости. Обнаружили также несколько пациентов с частичным дефицитом транслоказы и более легкой формой заболевания без вовлечения сердца. Определенных органических кислот в моче или плазме крови не обнаружили, хотя сообщается о повышенных уровнях длинноцепочечных ацилкарнитинов с длиной цепи С16-С18 в плазме.
Диагноз можно подтвердить с помощью генетического исследования. Функциональную активность карнитинацилкарнитинтранслоказы можно определить в культуре фибробластов или лимфобластов. Лечение такое же, как при нарушениях окисления др. длинноцепочечных жирных кислот.
4. Дефицит карнитин-пальмитоилтрансферазы II. Описано три формы дефицита СРТ-II. Зарегистрировано несколько случаев тяжелых проявлений с летальным исходом у новорожденных. Они характеризовались выраженным дефицитом фермента и ранней смертью, сопровождались дисплазией почек, мальформациями ГМ и легкими аномалиями развития лица. Более легкие дефекты проявляются во взрослом возрасте эпизодическим рабдомиолизом. Первый эпизод возникает, как правило, не ранее старшего детского или молодого возраста. Приступам нередко предшествует длительная ФН. Во время приступов отмечаются ноющие мышечные боли и миоглобинурия, которая м.б. достаточно тяжелой, с развитием почечной недостаточности. Уровни КФК сыворотки повышаются до 5000-100 000 единиц/л. Гипогликемия не описана, однако голодание может провоцировать приступы миоглобинурии. При биопсии мышц обнаруживают повышенное отложение нейтральных жиров.
Во взрослом возрасте миопатическая форма дефицита СРТ-П связана с распространенной мутацией с.338С>Т, p.S113L. Эта мутация приводит к экспрессии термолабильного белка, который меняется в ответ на повышение температуры мышц во время ФН, что проявляется миопатией.
Третья, промежуточная форма дефицита СРТ-II манифестирует в грудном или раннем детском возрасте и проявляется печеночной недостаточностью, кардиомиопатией и миопатией скелетных мышц с гипокетотической гипогликемией, спровоцированной голоданием. Для этой формы не характерны тяжелые аномалии развития, которые наблюдаются при форме с летальным исходом у новорожденных. Паттерн этой формы сходен с паттерном, наблюдаемым при дефиците VLCAD. Лечение идентичное.
Диагноз при всех формах дефицита СРТ-II можно установить с помощью молекулярно-генетического анализа в сочетании с обнаружением недостаточной активности фермента в мышечной или др. ткани или в культуре фибробластов.
в) Дефекты пути переноса электронов:
1. Дефицит электронпереносящего флавопротеина и дегидрогеназы электронпереносящего флавопротеина (глутаровая ацидемия 2-го типа, множественные дефекты дегидрирования ацил-КоА). Функция ETF и ETF-DH заключается в переносе электронов, полученных в результате реакций дегидрирования, в митохондриальную цепь переноса электронов. Катализаторами в этих реакциях дегидрирования выступают VLCAD, MCAD и SCAD, а также глутарил-КоА-дегидрогеназа и четыре фермента, участвующих в окислении аминокислот с разветвленной цепью (ВСАА; англ. branched-chain amino acids). Дефицит ETF или ETF-DH приводит к заболеванию, в котором сочетаются признаки нарушенного окисления жирных кислот и нарушенного окисления некоторых аминокислот. Полный дефицит любого белка в период новорожденности проявляется тяжелым заболеванием.
Для этого заболевания характерны ацидоз, гипокетотическая гипогликемия, кома, гипотония, кардиомиопатия и необычный запах вспотевших ног, который связан с ингибированием дегидрогеназы изовалерил-КоА. У некоторых новорожденных, страдающих такими дефектами, имеется врожденный дисморфизм лица и поликистоз почек, как при тяжелом дефиците СРТ-II.Это указывает на возможность токсического воздействия накопленных метаболитов в период в/утробного развития.
Диагноз можно установить путем определения ацилкарнитинового профиля и профиля органических кислот мочи в рамках неонатального скрининга методом сухого пятна. Оба исследования показывают наличие отклонений, соответствующих блокированию окисления жирных кислот (этилмалоната и С6-С10-дикарбоновых кислот), лизина (глутарата) и ВСАА (изовалерил-, изобутирил- и а-метилбутирил-глицина). Диагноз можно подтвердить путем генетического исследования на ETF (два гена — А и В) и ETF-дегидрогеназу. Большинство новорожденных с тяжелым поражением не переживают неонатальный период.
Частичный дефицит ETF и ETF-DH приводит к расстройству, которое может имитировать дефицит MCAD, или к более легким нарушениям окисления жирных кислот. У таких пациентов могут возникать приступы гипо-кетотической комы, индуцированной голоданием. В профиле органических кислот мочи наблюдают первичное повышение уровней дикарбоновых кислот и этилмалоната, которые являются промежуточными метаболитами короткоцепочечных жирных кислот. Имеется вторичный дефицит карнитина. У некоторых пациентов с легкими формами дефицита ETF/ETF-DH м.б. эффективно лечение высокими дозами рибофлавина — предшественника разл. флавопротеинов, участвующих в переносе электронов.
г) Дефекты пути синтеза кетонов. На заключительных этапах синтеза кетонов в процессе митохондриального β-окисления жирных кислот ацетил-КоА превращается в ацетоацетат с участием двух ферментов пути ГМГ-КоА (рис. 2).
1. Дефицит β-гидрокси-β-метилглутарил-КоА-синтазы. Этап с участием ГМГ-КоА-синтазы ограничивает скорость превращения ацетил-КоА, полученного в результате β-окисления жирных кислот в печени, в кетоны. Выявлено несколько пациентов с таким дефектом. Нарушение проявляется одной из форм гипокетотической гипогликемии натощак, без признаков нарушения функции сердца или мышц. Профиль органических кислот мочи демонстрирует только неспецифическую гипокетотическую дикарбоновую ацидурию. Уровни карнитина в плазме и тканях не отклоняются от нормы, в отличие от всех остальных форм нарушения окисления жирных кислот. Отдельный фермент синтаза, присутствующий в цитозоле и участвующий в биосинтезе ХС, не вовлекается в патологический процесс. Дефект ГМГ-КоА-синтазы экспрессируется только в печени (и почках). Его невозможно определить в культуре фибробластов. Диагноз можно установить методом генетического мутационного анализа. Избегание голодания, как правило, является эффективным методом лечения.
2. Дефицит β-гидрокси-β-метилглутарил-КоА (3-гидрокси-3-метилглутариловой ацидурии). См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
д) Нарушение утилизации кетоновых тел. Кетоновые тела, β-гидроксибутират и ацетоацетат, являются конечными продуктами окисления жирных кислот в печени. Они играют важную роль в качестве метаболического топлива для ГМ в период голодания. Три дефекта утилизации кетонов в ГМ и периферических тканях проявляются эпизодами гиперкетотической комы с гипогликемией или без нее.
1. Дефицит монокарбоксилатного транспортера-1. Описано ок. 10 пациентов с рецидивирующими приступами потенциально летального кетоацидоза с гипогликемией или без гипогликемии, вызванных дефицитом МСТ1. Этот переносчик встроен в плазматическую мембрану и кодируется геном SLC16A1. Он переносит кетоны из плазмы крови в ткани. Хотя в первых выявленных случаях пациенты были гомозиготными по инактивирующим мутациям МСТ1, заболевание может проявляться и у гетерозиготных носителей. У пациентов развивается тяжелый кетоацидоз, который провоцируется голоданием или инфекциями, перенесенными на первом году жизни. Гипогликемия присутствует не всегда. Проводят ДД с кетотической гипогликемией, которая наблюдается при более легких формах гликогеноза, напр. при дефиците фосфорилазы или фосфорилазы киназы. Лечение острых приступов включает в/в введение декстрозы («Глюкозы») с целью подавления липолиза и продолжающегося кетогенеза. Долгосрочная терапия включает избегание длительного голодания.
Диагноз можно заподозрить по необычно выраженному кетозу и по отсроченному снижению уровня кетонов после начала введения декстрозы («Глюкозы»). Специфических метаболических маркеров или методов, входящих в неонатальный скрининг, не существует. Диагноз можно установить путем генетического секвенирования SLC16A1.
2. Дефицит сукцинил-КоА-3-кетоацид-КоА-трансферазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше. Зарегистрировано несколько пациентов с дефицитом сукцинил-КоА-3-кетоацид-КоА-трансферазы (SCOT). Заболевание, как правило, проявляется рецидивирующими приступами тяжелого кетоацидоза у детей грудного возраста, провоцируемыми голоданием. Отклонения уровней ацилкарнитина плазмы и органических кислот мочи не позволяют отличить дефицит SCOT от др. причин кетоацидоза. Лечение приступов требует введения в/в декстрозы («Глюкозы») и больших количеств бикарбоната до стабилизации метаболического статуса. У пациентов, как правило, отмечается несоответствующая гиперкетонемия даже между приступами заболевания. SCOT отвечает за активацию ацетоацетата в периферических тканях, а сукцинил-КоА играет роль донора для образования ацетоацетил-КоА. Недостаточную активность фермента можно обнаружить в образцах клеток ГМ, мышц и фибробластов пациентов, страдающих этим заболеванием. Ген клонирован, и его большое количество мутаций описаны.
3. Дефицит β-кетотиолазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.