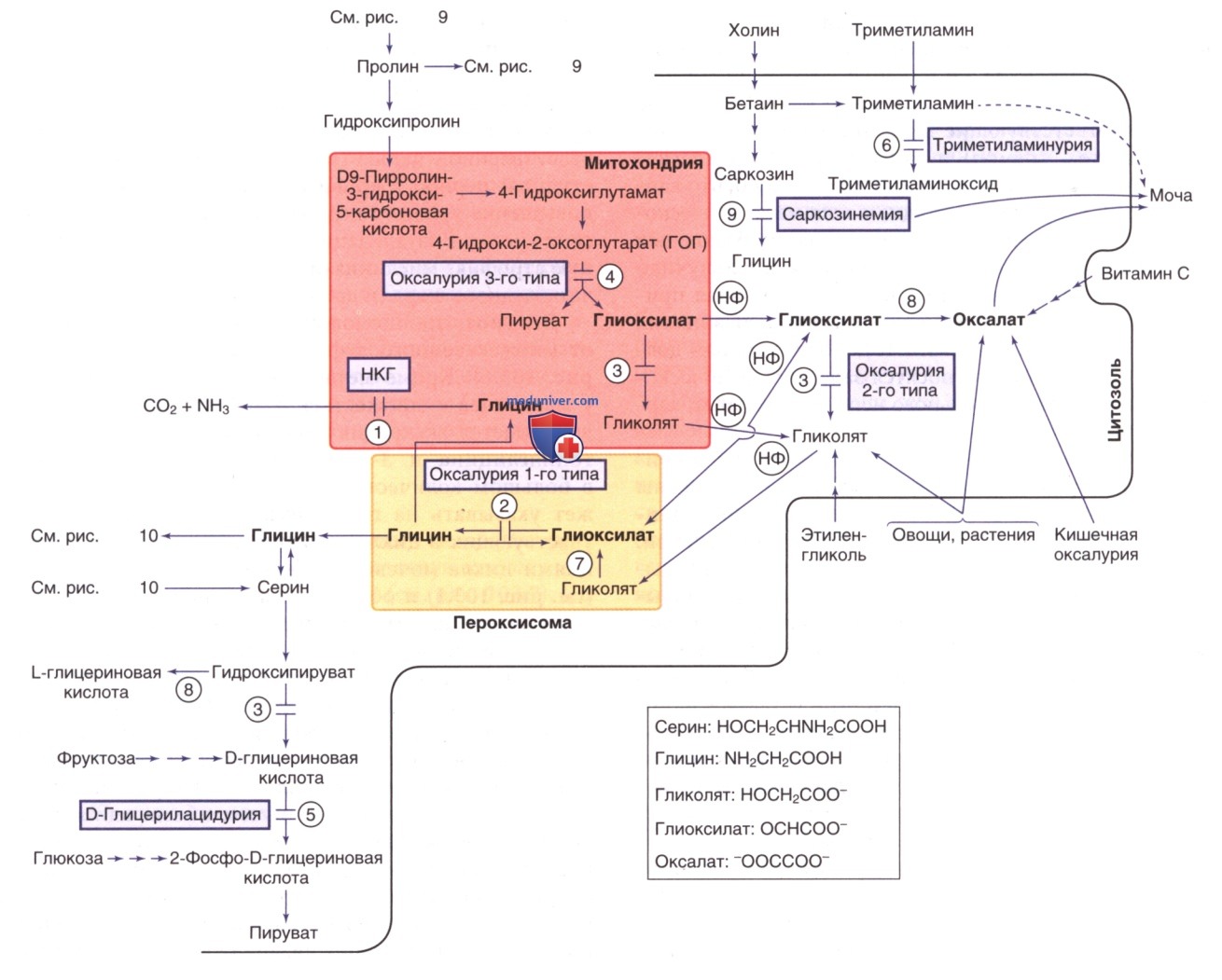
Глицин — заменимая аминокислота, синтезируемая в основном из серина и треонина. По структуре это простейшая аминокислота. Глицин участвует во многих реакциях организма, особенно в НС, где он действует как нейромедиатор (возбуждающий в коре ГМ, ингибирующий в стволе ГМ и спинном мозге). Для его основного катаболического пути требуется система расщепления глицина, пиридоксальфосфат-зависимый комплекс митохондриальных ферментов, который превращает глицин в диоксид углерода и аммиак и переводит α-углерод в тетрагидрофолат (см. рис. 8).
Система расщепления глицина состоит из четырех белков: белка Р (глициндекарбоксилаза), белка Н, белка Т и белка L, которые кодируются четырьмя разными генами.
а) Гипоглицинемия. Дефекты пути биосинтеза серина способствуют развитию дефицита глицина в сочетании с дефицитом серина в физиол. жидкостях, особенно в СМЖ. Случаи изолированного первичного дефицита глицина не зарегистрированы.
б) Гиперглицинемия. Повышенное содержание глицина в физиол. жидкостях возникает при пропионовой, метилмалоновой, изо-валериановой ацидемиях и дефиците β-кетотиолазы, которые в совокупности называются кетотической гиперглицинемией из-за сосуществования ацидоза и кетоза. Патогенез гиперглицинемии при этих нарушениях до конца не изучен, но было показано, что у некоторых пациентов наблюдается ингибирование системы ферментов расщепления глицина разл. органическими кислотами.
Термин некетотическая гиперглицинемия (НКГ) применяется для обозначения клинического состояния, вызванного генетическим дефицитом ферментной системы расщепления глицина (см. рис. 8). При этом заболевании гиперглицинемия возникает при отсутствии кетоза.
в) Некетотическая гиперглицинемия (глициновая энцефалопатия). Выявлены четыре формы НКГ: неонатальная, младенческая, с поздним началом и транзиторная.
1. Неонатальная некетотическая гиперглицинемия. Это наиболее часто встречаемая форма НКГ. Клинические проявления развиваются в первые несколько дней жизни (между 6 ч и 8 сут после рождения). Развиваются отказ от груди, вялое сосание, заторможенность и тяжелая гипотония, которые могут быстро прогрессировать до глубокой комы, апноэ и смерти. Часто наблюдаются судороги, особенно миоклонические приступы и икота.
Лабораторные исследования выявляют гиперглицинемию умеренной и тяжелой степени (в 8 раз превышающую норму) и гиперглицинурию. Диагностическими признаками НКГ являются явное повышение концентрации глицина в СМЖ (в 15-30 раз от нормы) и высокое соотношение концентрации глицина в СМЖ и плазме (значение >0,08 при норме <0,02). У пациентов с этой патологией pH крови обычно в норме, а в моче отсутствуют органические кислоты. Содержание серина в СМЖ м.б. низким.
Примерно 30% младенцев с НКГ умирают, несмотря на поддерживающую терапию. У выживших детей развивается глубокая задержка психомоторного развития и трудноизлечимые судорожные расстройства (миоклонические судороги и/или большие эпилептические припадки). У некоторых выживших отмечались гидроцефалия, требующая шунтирования, и легочная гипертензия. У некоторых пациентов отмечена транзиторная гипергликемия.
2. Младенческая некетотическая гиперглицинемия. Для младенческой НКГ признаки и симптомы, характерные для неонатальной НКГ, возникают в возрасте >6 мес. Частыми признаками являются судороги и гипотония. Младенческая НКГ, вероятно, является более легкой формой неонатальной НКГ; младенцы обычно выживают, при этом наблюдается не столь серьезная умственная отсталость, как при неонатальной форме.
Лабораторные признаки у пациентов с младенческой НКГ идентичны таковым при неонатальной форме.
3. Некетотическая гиперглицинемия с поздним началом. К клиническим проявлениям этой атипичной формы НКГ относятся прогрессирующая спастическая диплегия, атрофия зрительного нерва и хореоатетоидные движения. Возраст начала заболевания 2-33 года. У некоторых пациентов при интеркуррентной инфекции могут эпизодически возникать симптомы делирия, хореи и паралича вертикального взора. Психическое развитие обычно в норме. У некоторых пациентов отмечались легкие когнитивные нарушения и нечастые судороги.
Лабораторные признаки НКГ с поздним началом похожи на симптомы НКГ новорожденных, но выражены слабее.
Все формы НКГ следует отличать от кетотической гиперглицинемии, дефицита пиридокс(ам)инфосфатоксидазы, воздействия вальпроевой кислоты и временной глициновой энцефалопатии. Вальпроевая кислота может умеренно повышать концентрацию глицина в крови, СМЖ и моче. Повторные анализы после отмены ЛП способствуют постановке диагноза.
4. Транзиторная некетотическая гиперглицинемия. Большинство клинических и лабораторных проявлений транзиторной НКГ неотличимы от неонатальной формы. Однако в 2-8 нед может наступить полное клиническое выздоровление, а повышенные уровни глицина в плазме и СМЖ нормализуются после прекращения приема глициноснижающих ЛП. У некоторых пациентов развитие соответствует норме, без неврологических последствий, у других же отмечается умственная отсталость. Этиология этого заболевания неизвестна, но считается, что это следствие незрелости ферментной системы.
5. Диагностика и лечение. Диагноз НКГ можно заподозрить при повышенном содержании глицина в плазме/СМЖ и аномальном соотношении глицина в СМЖ и плазме. Диагноз подтверждают молекулярным анализом генов, обусловливающих развитие НКГ. В редких случаях для установления диагноза требуется анализ фермента в образцах печени/ГМ. Активность ферментов в неонатальной форме близка к нулю, тогда как в др. формах присутствует некоторая остаточная активность. У большинства пациентов с неонатальным НКГ дефект фермента находится в белке Р (75%); дефекты в Т-белке составляют 20% случаев, тогда как <1% вызваны патогенными вариантами в Н-белке.
Эффективного лечения не разработано. Заместительное переливание крови, ограничение глицина в питании и введение натрия бензоата/фолата не повлияли на неврологическую симптоматику при тяжелых формах НКГ. У пациентов с легкой формой НКГ после приема натрия бензоата внутрь может наблюдаться клиническое улучшение. ЛП, защищающие нейроны от влияния глицина (напр., декстрометорфан и фелбамат), помогают лишь при легких формах данного состояния.
НКГ наследуется по АуР-типу. Частота встречаемости неизвестна, но высокая частота заболевания была отмечена в северной Финляндии (1:12 000 живорожденных), что позволяет предположить, что это заболевание, вероятно, не диагностируется. Ген белка Р (GLDC) находится в хромосоме 9р24.1. Ген, кодирующий белок Т (АМТ), находится в хромосоме 3р21.31, а ген белка Н (GCSH) расположен в хромосоме 16q23.2. Ген L-белка (DLD) в хромосоме 7q31.7 кодирует дигидролипоамиддегидрогеназу, компонент ЕЗа-кетокислотных дегидрогеназных комплексов.
Данный ген обсуждается в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше. Пренатальную диагностику выполняют путем выявления известных семейных патогенных вариантов в дефектном гене/путем проведения анализа активности фермента в биоптатах ворсинок хориона.
г) Саркозинемия. В крови и моче наблюдаются повышенные концентрации саркозина (N-метилглицина), тем не менее саркозинемия не имеет постоянной клинической картины. Это АуР-метаболическое нарушение вызвано дефектом саркозиндегидрогеназы (фермента, превращающего саркозин в глицин) (см. рис. 8). Ген этого фермента (SARDH) находится в хромосоме 9q34.2.
д) Первичная триметиламинурия. Триметиламин обычно образуется в кишечнике при бактериальном расщеплении холина и оксида триметиламина. Основными источниками холина являются яичный желток и печень, а основным источником оксида триметиламина — рыба. Триметиламин абсорбируется и окисляется в печени триметиламиноксидазой (флавинсодержащими монооксигеназами) до триметиламиноксида, который не имеет запаха и выводится с мочой (см. рис. 8).
Дефицит указанного фермента приводит к значительной экскреции триметиламина с мочой. Исходящий от тела больных сильный запах тухлой рыбы может создавать серьезные социальные и психосоциальные проблемы. У ЗЛ после употребления большого количества указанных выше продуктов может возникать временная симптоматическая триметиламинурия. Лечение в виде приема внутрь активированного угля, коротких курсов приема внутрь метронидазола, неомицина и лактулозы временно уменьшают запах тела.
Значительно снижает запах ограничение в рационе рыбы, яиц, печени и др. источников холина (напр., орехов, злаков). Местное использование кислого мыла (pH 5,5) также может помочь контролировать запах. Ген триметиламиноксидазы (FMO3) расположен в хромосоме 1q24.3.
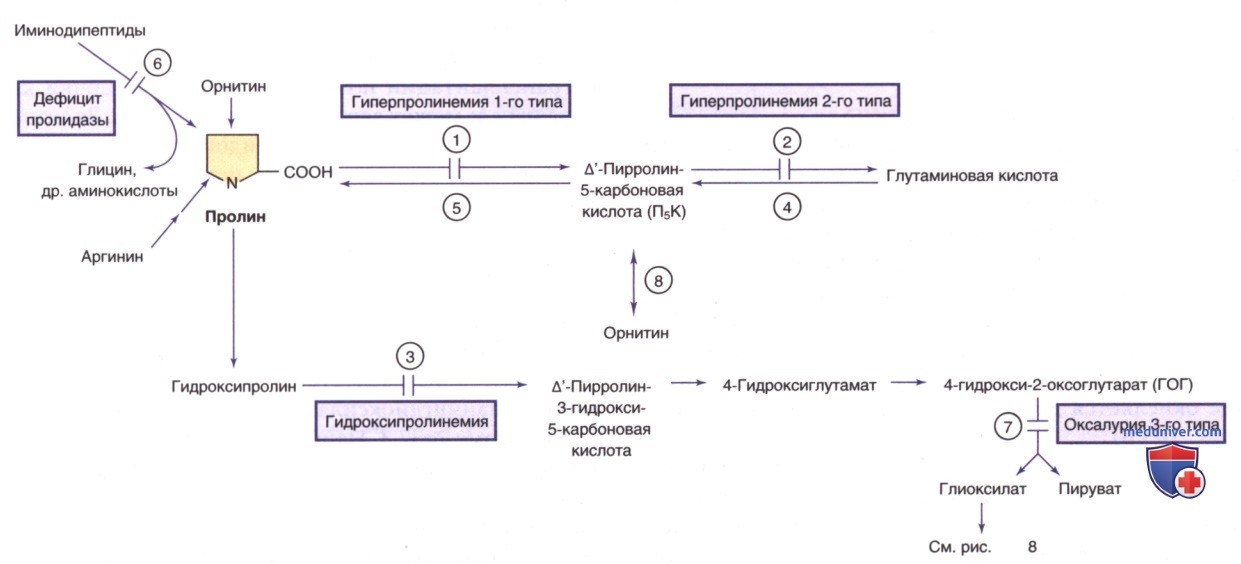
е) Гипероксалурия и оксалоз. В норме щавелевая кислота образуется преимущественно при окислении глиоксиловой кислоты и, в меньшей степени, при окислении аскорбиновой кислоты (см. рис. 8). Глиоксиловая кислота образуется в результате окисления гликолевой кислоты и глицина в пероксисомах и катаболизма гидроксипролина в митохондриях (см. рис. 9). Овощи и продукты, содержащие щавелевую кислоту (напр., шпинат, ревень и миндальное молоко), являются основными экзогенными источниками гликолевой и щавелевой кислот; в большей степени глиоксиловая и щавелевая кислоты образуются эндогенно.
Обычно большая часть глиоксилата, производимого в организме, транспортируется в пероксисомы, где он превращается в глицин под действием фермента аланин: глиоксилатаминотрансферазы. Дефицит этого фермента приводит к гипероксалурии 1-го типа. Под действием фермента глиоксилатредуктазы/гидроксипируватредук-тазы большая часть остающегося глиоксилата в цитозоле восстанавливается до гликолата. Дефицит этого фермента приводит к гипероксалурии 2-го типа. Эти два пути защищают организм от чрезмерного продуцирования щавелевой кислоты (см. рис. 8).
Любой глиоксилат, который не м.б. использован указанными путями, легко превращается в щавелевую кислоту под действием фермента ЛДГ. Щавелевая кислота не может участвовать в дальнейшем метаболизме человека и выводится с мочой в виде оксалатов. Оксалат кальция относительно нерастворим в воде и осаждается в тканях (почках и суставах) при увеличении его концентрации в организме.
Вторичная гипероксалурия развивалась при недостаточности пиридоксина (кофактор аланин: глиоксилатаминотрансферазы), у пациентов с ВЗК, обширной резекцией тонкой кишки/брыжеечной части тонкой кишки (кишечная гипероксалурия), после приема этиленгликоля/высоких доз витамина С и после введения анестетика метоксифлурана (который непосредственно окисляется до щавелевой кислоты). После употребления в пищу растительных продуктов с высоким содержанием щавелевой кислоты (напр., щавеля)/преднамеренного приема щавелевой кислоты может развиться острая гипероксалурия, способная оказаться смертельной.
Осаждение оксалата кальция в тканях вызывает гипокальциемию, некроз печени, почечную недостаточность, сердечную аритмию и приводит к летальному исходу. По оценкам, летальная доза щавелевой кислоты составляет 5-30 г.
Первичная гипероксалурия представляет собой группу заболеваний, при которых в организме накапливается большое количество оксалатов. На сегодняшний день установлено три типа первичной гипероксалурии. Термин оксалоз отражает отложение оксалата кальция в паренхиматозных тканях.
1. Первичная гипероксалурия 1-го типа. Это редкое нарушение (с частотой встречаемости 1:120 000 живорожденных в Европе) является наиболее распространенной формой первичной гипероксалурии. Заболевание вызвано дефицитом пероксисомального фермента аланин: глиоксилатаминотрансферазы, который экспрессируется только в пероксисомах печени и требует пиридоксина (витамина В6) в качестве кофактора. При отсутствии этого фермента глиоксиловая кислота не м.б. преобразована в глицин и поэтому переносится в цитозоль, где окисляется до щавелевой кислоты (см. выше, а также рис. 8).
Симптомы проявляются в разном возрасте: от новорожденных до взрослых пациентов. У большинства пациентов симптомы проявляются в старшем детском/раннем подростковом возрасте. В 20% случаев симптомы развиваются <1 года. Первоначальные клинические проявления обусловлены МКБ и нефрокальцинозом. Почечная колика и бессимптомная гематурия приводят к постепенному снижению функции почек, что проявляется задержкой роста и уремией. При отсутствии лечения большинство пациентов умирают в возрасте <20 лет от почечной недостаточности.
К др. частым проявлениям заболевания относятся задержка физического развития, низкий рост, кальциноз артерий, аритмия, СН, гипотиреоз и узелки на коже. Острый артрит встречается редко, и его можно ошибочно принять за подагру, поскольку у пациентов с гипероксалурией 1-го типа уровень мочевой кислоты обычно повышен. У нескольких пациентов наблюдались кристаллическая ретинопатия и оптическая невропатия, приводящие к потере зрения.
Наиболее важным лабораторным признаком является существенное увеличение экскреции оксалатов с мочой (нормальная экскреция составляет 10-50 мг/сут). Наличие кристаллов оксалата в осадке мочи малоинформативно, поскольку такие кристаллы наблюдаются в моче ЗЛ. У большинства пациентов (но не у всех) происходит экскреция гликолевой и глиоксиловой кислот с мочой. Диагноз м.б. подтвержден путем идентификации патогенных вариантов в гене AGXT/путем проведения ферментативного анализа в образцах печени.
Лечение направлено на снижение продуцирования щавелевой кислоты и повышения экскреции оксалата кальция. Пациентам с первичной гипероксалурией 1-го типа необходимо пройти трехмесячный курс лечения пиридоксином с целью установления чувствительности к этому ЛП. У 30% пациентов (напр., гомозиготных по патогенному варианту c.508G>A в AGXT) прием больших доз пиридоксина снижает уровень оксалата в плазме и экскрецию оксалата с мочой. Для увеличения удаления оксалата кальция и профилактики МКБ рекомендуются употребление большого количества жидкости (2-3 л/м2 в сутки с контролем водного баланса), алкалинизация мочи, прием фосфатных БАД, мониторинг потребления витаминов С и D и исключение ЛП, увеличивающих экскрецию кальция с мочой (напр., петлевые диуретики).
МКБ должны лечить опытные урологи, поскольку чрезмерная хирургическая травма может способствовать развитию дисфункции почек. У некоторых пациентов (напр., при подготовке к трансплантации/если трансплантация не показана) используются стратегии замещения функции почек (напр., гемодиализ).
В качестве наиболее радикального метода лечения применяют трансплантацию органов. Принятие решения о трансплантации почки/печени/печени-почки является сложным и может зависеть от мед. центра. За исключением взрослых пациентов с пиридоксин-зависимой формой заболевания, трансплантация почки пациентам с почечной недостаточностью может оказаться неэффективной из-за повторного оксалоза в трансплантированной почке. Комбинированная трансплантация печени и почек позволяет значительно снизить уровень оксалатов в плазме и моче и считается более эффективной стратегией лечения этого заболевания, особенно у детей.
Заболевание наследуется по АуР-типу. Ген этого фермента (аланинглиоксилатаминотрансферазы) расположен в хромосоме 2q37.3. Наиболее частая мутация у пациентов с высокой остаточной активностью фермента (c.508G>A, p.Glyl70Arg) приводит к ошибочной экспрессии фермента в митохондриях вместо пероксисом и потере функции in vivo. Пренатальную диагностику выполняют путем ДНК-анализа образцов ворсинок хориона/измерения активности ферментов печени плода в образце, полученном игольной биопсией.
2. Первичная гипероксалурия 2-го типа (L-глицериновая ацидурия). Причиной этого редкого заболевания является дефицит ферментного комплекса глиоксилатредуктазы/ги-дроксипируватредуктазы (см. рис. 8). Недостаточная активность этого комплекса приводит к накоплению двух промежуточных метаболитов — гидроксипирувата (кетокислотное производное серина) и глиоксиловой кислоты. Оба указанных соединения далее метаболизируются с помощью ЛДГ до L-глицериновой кислоты и щавелевой кислоты соответственно. Высокая частота распространенности этого нарушения зарегистрирована у племени индейцев западного Оджибва из Манитобы.
Первичная гипероксалурия 2-го типа приводит к отложению оксалата кальция в почечной паренхиме и МВП. В возрасте <2 лет могут появиться камни в почках, сопровождающиеся почечной коликой и гематурией. Почечная недостаточность при этом заболевании встречается реже, чем при первичной гипероксалурии 1-го типа.
В моче отмечается большое количество L-глицериновой кислоты вдобавок к высокому уровню оксалата. L-Глицериновая кислота в моче считается патогномоничным признаком при первичной гипероксалурии 2-го типа. Экскреция с мочой гликолевой и глиоксиловой кислот не повышена. Наличие в моче L-глицериновой кислоты без повышенного уровня гликолевой и глиоксиловой кислот является отличительным признаком гипероксалурии 2-го типа от гипероксалурии 1-го типа. Диагноз подтверждают молекулярным анализом GRHPR (9р13.2)/ферментным анализом при биопсии печени.
Принципы терапии те же, что и при первичной гипероксалурии 1-го типа. Некоторым пациентам выполняют трансплантацию почки; опыта трансплантации почки и печени в настоящее время нет.
3. Первичная гипероксалурия 3-го типа. У 10% пациентов с первичной гипероксалурией наблюдается дефицит 4-гидрокси-2-оксоглутаратальдолазы 1, что является основной причиной гипероксалурии 3-го типа. Фермент кодируется геном HOGA1, расположенным в хромосоме 10q24.2. Указанный митохондриальный фермент катализирует заключительный этап метаболического пути гидроксипролина, продуцирующего пируват и глиоксилат из 4-гидрокси-2-оксоглутарата (см. рис. 8, 9).
Исследования in vitro показывают, что 4-гидрокси-2-оксоглутарат, накапливающийся у пациентов с гипероксалурией 3-го типа, при высоких концентрациях ингибирует активность фермента глиоксилатредуктазы/гидроксипируватредуктазы. Это ингибирование приводит к формированию биохим. фенотипа, подобно гипероксалурии 2-го типа (см. рис. 8).
У пациентов с первичной гипероксалурией 3-го типа в раннем детстве обычно присутствовали камни в почках из оксалата кальция, а также было выявлено бессимптомное заболевание у старших братьев/сестер. Функция почек может постепенно снижаться, что в некоторых случаях приводит к терминальной стадии почечной недостаточности. Отличительным признаком этого заболевания являются повышенные концентрации 4-гидрокси-2-оксоглутарата в моче, сыворотке и биоптатах печени пациентов. Лечение включает обильное питье, прием цитрата/фосфата внутрь, что препятствует образованию в почках камней из оксалата кальция, а также профилактику обезвоживания для предотвращения ОПН. При тяжелых формах этого нарушения для лечения терминальной стадии почечной недостаточности могут потребоваться диализ и трансплантация.
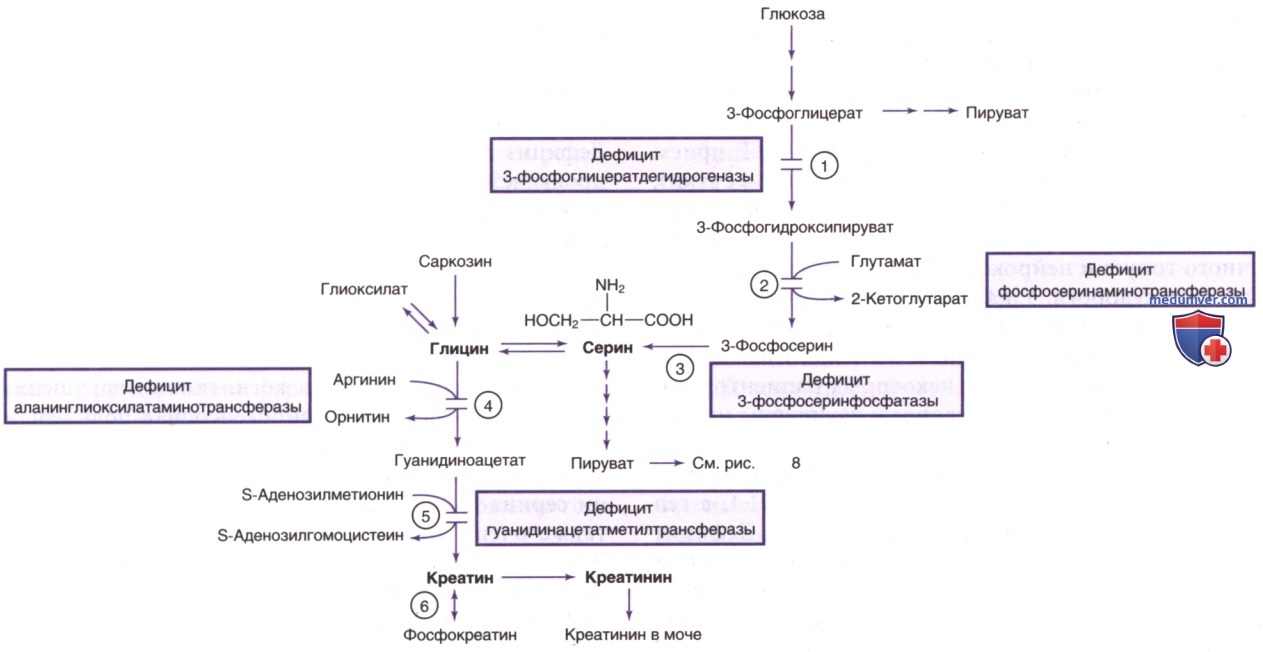
4. Нарушения, обусловленные дефицитом креатина. Креатин синтезируется из аргинина и глицина преимущественно в печени, ПЖЖ и почках и, в меньшей степени, в ГМ. Затем креатин транспортируется в мышцы и ГМ, где наблюдается высокая активность фермента КФК (рис. 10). Фосфорилирование и дефосфорилирование креатина в сочетании с АТФ и АДФ обеспечивают высокоэнергетические реакции транспорта фосфата в этих органах. Креатин неферментативно метаболизируется до креатинина с постоянной суточной скоростью и выводится с мочой.
Известны три генетических нарушения, которые вызывают дефицит креатина в ГМ и др. тканях. В биосинтезе креатина участвуют два фермента: аргинин-глицинамидинотрансферазы (АГАТ) и гуанидинацетат-метилтрансфераза (ГАМТ; рис. 10). Оба заболевания могут поддаваться лечению при приеме креатина, особенно когда лечение начинают в раннем возрасте. Третье нарушение, Х-сцепленный наследственный дефект, вызвано дефицитом транспортного белка креатинина, опосредующего поглощение креатина ГМ и мышцами.
Для всех трех нарушений характерна сходная клиническая картина. Уже с первых недель/месяцев жизни появляются симптомы, обусловленные нарушением функций ГМ и мышц. Часто наблюдаются задержка психического развития, умственная отсталость, задержка речи, психиатрические симптомы (аутизм и психоз), гипотония, атаксия и судороги. При дефиците ГАМТ и транспортного белка креатинина отмечались дистонические движения.
К лабораторным признакам относятся снижение креатина в плазме у пациентов с дефектами АГАТ и ГАМТ. Для диагностики указанных нарушений недостаточно определить только содержание креатина в плазме. Кроме нарушения реабсорбции креатина в почках, у пациентов мужского пола с нарушением транспортного белка креатинина повышается соотношение креатина и креатинина в моче. У носителей-женщин также м.б. слегка повышен этот показатель. ДК нарушения ГАМТ является заметное повышение уровня гуанидиноацетата в крови, моче и особенно в СМЖ. При нарушении АГАТ, наоборот, в физиол. жидкостях могут наблюдаться низкие уровни гуанидиноацетата.
Отсутствие креатина и креатинфосфата (при всех трех нарушениях) и высокий уровень гуанидиноацетата (при нарушении ГАМТ) в ГМ можно определить методом магнитно-резонансной спектроскопии. На снимках МРТ ГМ можно увидеть повышенную интенсивность сигнала в бледном шаре. Диагноз дефицита АГАТ/ГАМТ подтверждается анализом ДНК/измерением ферментативной активности в культуре фибробластов (ГАМТ) и лимфобластах (АГАТ). Диагноз дефицита транспортного белка креатинина подтверждается анализом ДНК/поглощения креатина фибробластами.
Результаты лечения зависят от возраста, при этом предпочтительно начинать лечение в неонатальном/досимптомном периоде. Прием креатина моногидрата внутрь (до 400-800 мг/кг в сутки) может снизить мышечную слабость у большинства пациентов с дефицитом АГАТ и в некоторых случаях улучшить нейрокогнитивное развитие.
У пациентов с дефицитом ГАМТ прием креатина моногидрата внутрь (до 400-800 мг/кг в сутки), орнитина внутрь (до 400-800 мг/кг в сутки) и ограничение аргинина в питании могут привести к улучшению мышечного тонуса и нейрокогнитивного развития, а также облегчить судороги. У пациентов с дефицитом транспортного белка креатинина принимаемые БАД креатина и его предшественников (аргинин и глицин) не восстанавливают креатин в ГМ, но у некоторых пациентов может наблюдаться облегчение судорог и улучшение нейрокогнитивного развития.
Дефекты АГАТ и ГАМТ наследуются как АуР-признаки. Ген АГАТ (GATM) находится в хромосоме 15q21.1, а ген ГАМТ (GAMT) расположен в хромосоме 19р13.3. Дефицит транспортного белка креатинина представляет собой Х-сцепленное заболевание, а ген (SLC6A8) находится в Xq28. Это последнее нарушение является наиболее частой причиной дефицита креатина, на его долю приходится до 1-2% умственной отсталости у мужчин по неизвестной причине.