N-ацетиласпарагиновая кислота, производное аспарагиновой кислоты, синтезируется в ГМ и присутствует в высокой концентрации аналогично глутаминовой кислоте. Исследования показывают, что N-ацетиласпарагиновая кислота выполняет несколько функций, напр. обеспечивает запас ацетата для синтеза миелина и является органическим осмолитом, который помогает регулировать осмоляльность ГМ. Однако ее функции изучены недостаточно.
Аспартоацилаза отщепляет N-ацетильную группу от N-ацетиласпарагиновой кислоты. Дефицит аспартоацилазы приводит к развитию болезни Канавана (Canavan), тяжелой лейкодистрофии, характеризующейся избыточной экскрецией N-ацетиласпарагиновой кислоты и губчатой дегенерацией белого в-ва ГМ. Болезнь Канавана является АуР-заболеванием, которое чаще встречается в популяции евреев-ашкенази, чем у представителей др. этнических групп. Частота рождений с данной патологией у евреев ашкенази составляет 1:6400-13 500.
В др. этнических группах частота встречаемости тяжелой формы болезни Канавана составляет 1:100 000 рождений. Дефектный ген болезни Канавана (ASPA) расположен на хромосоме 17. Пациентам, членам их семей и больным из групп риска м.б. рекомендовано генетическое тестирование.
а) Этиология и патология. Дефицит фермента аспартоацилазы приводит к накоплению N-ацетиласпарагиновой кислоты в ГМ, особенно в белом в-ве, и значительной экскреции этого соединения с мочой. В крови и СМЖ также повышено содержание этой кислоты. При биопсии ГМ пациентов с болезнью Канавана наблюдаются губчатая дегенерация миелиновых волокон, астроцитарный отек и удлиненные митохондрии. В белом в-ве отмечаются выраженная вакуолизация и набухание астроцитов.
Электронная микроскопия выявляет деформированные митохондрии. По мере прогрессирования заболевания желудочки увеличиваются вследствие церебральной атрофии.
б) Клинические проявления. Болезнь Канавана характеризуется широким спектром проявлений в зависимости от тяжести течения. Младенцы обычно выглядят здоровыми при рождении, поскольку заболевание может не проявляться до 3-6 мес, далее развиваются прогрессирующая макроцефалия, тяжелая гипотония, стойкое запрокидывание головы и задержка психомоторного развития.
По мере прогрессирования заболевания возникают спастичность, скованность суставов и контрактуры. Развиваются атрофия зрительного нерва и судороги. На первом году жизни могут возникать трудности с кормлением, отставание в прибавке МТ и ГЭР. Появляются проблемы с глотанием, может потребоваться назогастральное кормление/постоянная гастростомия. В прошлом большинство пациентов умирали в возрасте <10 лет. В настоящее время, благодаря развитию мед. технологий и улучшению поддерживающей терапии, такие дети часто доживают до 2-3-го десятилетия жизни.
в) Атипичная болезнь Канавана. Ювенильная/легкая форма болезни Канавана встречается реже, чем младенческая, и наиболее распространена среди евреев, не относящихся к ашкенази. Пациенты с ювенильной болезнью Канавана обычно страдают легкой задержкой речевого и моторного развития и могут иметь пигментный ретинит. Др. типичные признаки болезни Канавана обычно отсутствуют. У этих детей наблюдается умеренно повышенная экскреция N-ацетиласпарагиновой кислоты с мочой, что свидетельствует о болезни Канавана. На МРТ ГМ виден более интенсивный сигнал в базальных ганглиях, а не его тотальное поражение, что иногда приводит к ошибочному диагнозу митохондриального заболевания.
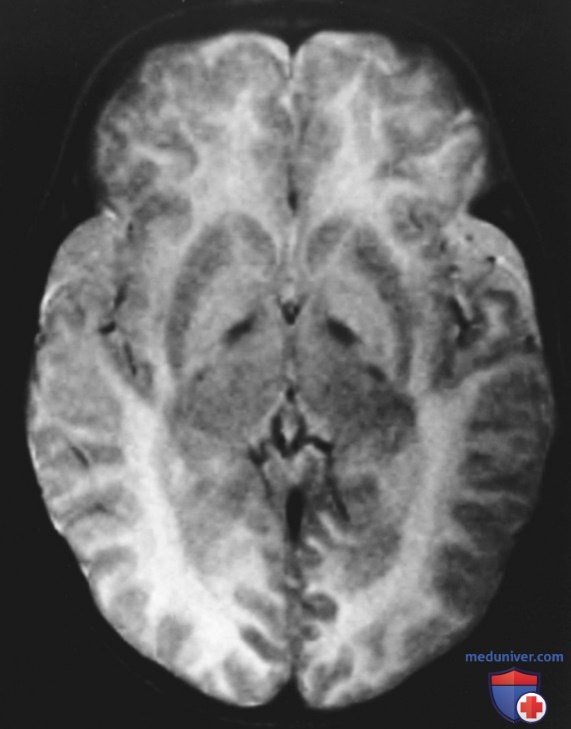
г) Диагностика. При типичном течении у пациента с болезнью Канавана при проведении КТ и МРТ выявляется диффузная дегенерация белого в-ва, преимущественно в полушариях ГМ, с меньшим вовлечением мозжечка и ствола (рис. ниже). Может потребоваться повторное обследование. Магнитно-резонансная спектроскопия, выполняемая во время МРТ, м.б. необходима для выявления высокого пика N-ацетиласпарагиновой кислоты, что позволяет предположить болезнь Канавана.
Аксиальная Т-взвешенная магнитно-резонансная томография двухлетнего пациента с болезнью Канавана. Видно существенное утолщение белого в-ва
Диагноз м.б. поставлен на основании повышения N-ацетиласпарагиновой кислоты в моче/крови. В моче ЗЛ эта кислота обнаруживается только в следовых количествах (24+16 мкмоль/ммоль креатинина), тогда как у пациентов с болезнью Канавана его концентрация находится в диапазоне 1440±873 мкмоль/ммоль креатинина. Высокое содержание N-ацетиласпарагиновой кислоты можно обнаружить в плазме, СМЖ и тканях ГМ. Для подтверждения диагноза часто определяют аспартоацилазу в фибробластах, но это не обязательно. Активность аспартоацилазы в фибробластах облигатных носителей составляет <50% активности, обнаруженной у ЗЛ.
У пациентов с болезнью Канавана всегда следует проводить генотипирование с определением мутации ASPA. ДД болезни Канавана должна включать болезнь Александера (Alexander), которая представляет собой др. лейкодистрофию, сопровождающуюся макроцефалией. Болезнь Александера развивается вследствие дефекта синтеза глиального фибриллярного кислого белка, и диагноз м.б. исключен при проведении молекулярной диагностики лимфоцитов крови.
Известны два преобладающих патогенных варианта, приводящих к болезни Канавана в популяции евреев-ашкенази. Первым вариантом является аминокислотная замена (Е285А), при котором глутаминовая кислота заменяется аланином. Эта мутация встречается наиболее часто и охватывает 83% из 100 мутантных аллелей, исследованных в популяции евреев-ашкенази. Вторым распространенным патогенным вариантом является замена тирозина на нонсенс-мутацию, приводящая к остановке кодирующей последовательности (Y231X). Этот вариант составляет 13% мутантных аллелей. У нееврейского населения наблюдаются более разнообразные патогенные варианты, и два вышеуказанных варианта встречаются редко.
Др. мутация (А305Е), замещение глутаминовой кислоты аланином, составляет 40% из 62 мутантных аллелей у нееврейских пациентов. Описано >50 патогенных вариантов, встречаемых в нееврейских популяциях. При болезни Канавана важно получить молекулярный диагноз, поскольку это позволит провести пренатальное консультирование и дать рекомендации семьи. Если мутации неизвестны, пренатальная диагностика основывается на концентрации N-ацетиласпарагиновой кислоты в околоплодных водах. У пациентов-евреев-ашкенази частота носителей может достигать 1:40, что близко к частоте встречаемости болезни Тея-Сакса (Тау-Sachs). Для еврейской популяции существует скрининг носителей болезни Канавана.
Корреляция генотипа, фенотипа и экспрессия аспартоацилазы показывают, что исследования экспрессии позволяют лучше понять механизмы болезни. Пациенты с ювенильными/легкими формами заболевания были сложными гетерозиготами с легким патогенным вариантом на одном и тяжелым на др. аллелях. Легкие варианты включают p.Tyr288Cys и p.Arg71His.
д) Лечение и профилактика. В настоящее время специфическое лечение не разработано. Недавние исследования генной терапии с использованием рекомбинантных аденоассоциированных вирусов показали некоторые «+» результаты при лечении мышей с отключенным геном, но клинические исследования на людях еще не проводились. Проблемы с питанием и судороги следует лечить индивидуально. Генетическое консультирование, тест на носительство и пренатальная диагностика являются единственными методами профилактики. Попытки генной терапии у детей с болезнью Канавана показали отсутствие долгосрочных побочных явлений, некоторое снижение уровня N-ацетиласпарагиновой кислоты в ГМ, меньшую частоту приступов и стабилизацию общего клинического состояния.