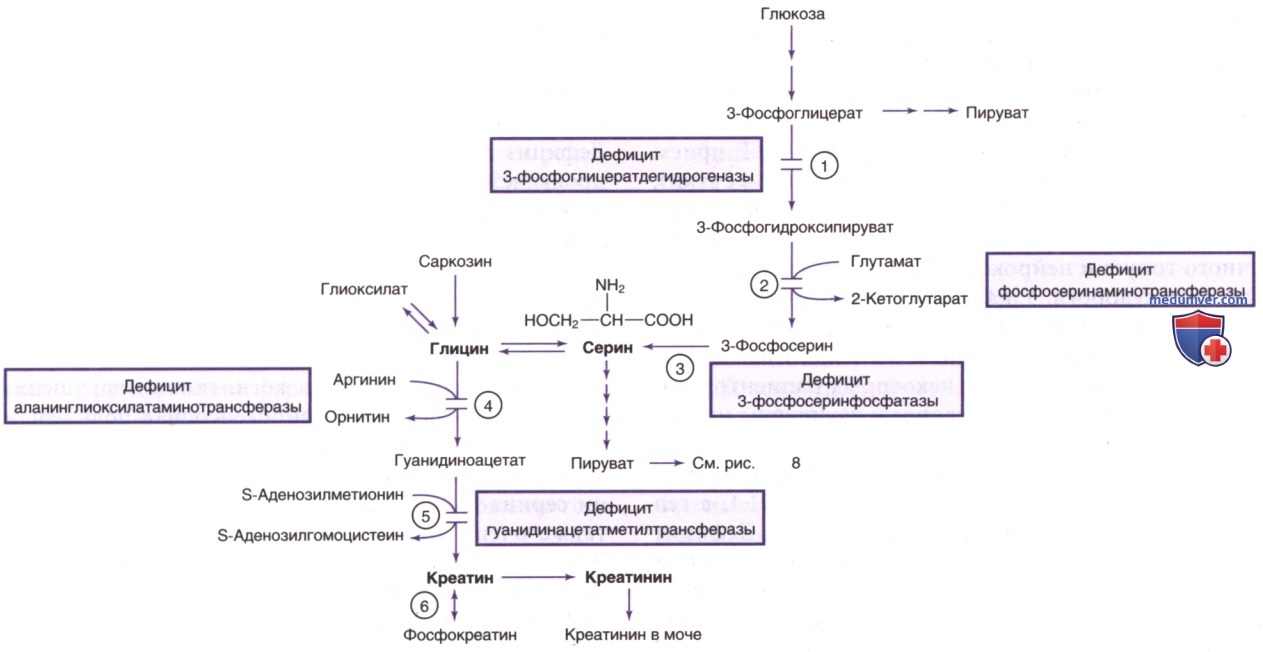
Серин представляет собой заменимую аминокислоту, поступающую из пищевых источников и продуцируемую путем эндогенного синтеза, в основном из глюкозы и глицина. Эндогенное продуцирование серина составляет важную часть суточной потребности в этой аминокислоте, особенно в синапсах, где он способствует метаболизму фосфолипидов, а также D-серина и глицина, участвующих в нейропередаче.
Следовательно, дефицит любого из ферментов, участвующих в биосинтезе серина/его транспорте, приводит к развитию неврологической симптоматики.
С дефицитом серина связан широкий спектр клинических проявлений, который варьирует от синдрома Ноя-Лаксовой (Neu-Laxova) при тяжелых формах до эпилепсии и задержки психического развития при легких формах.
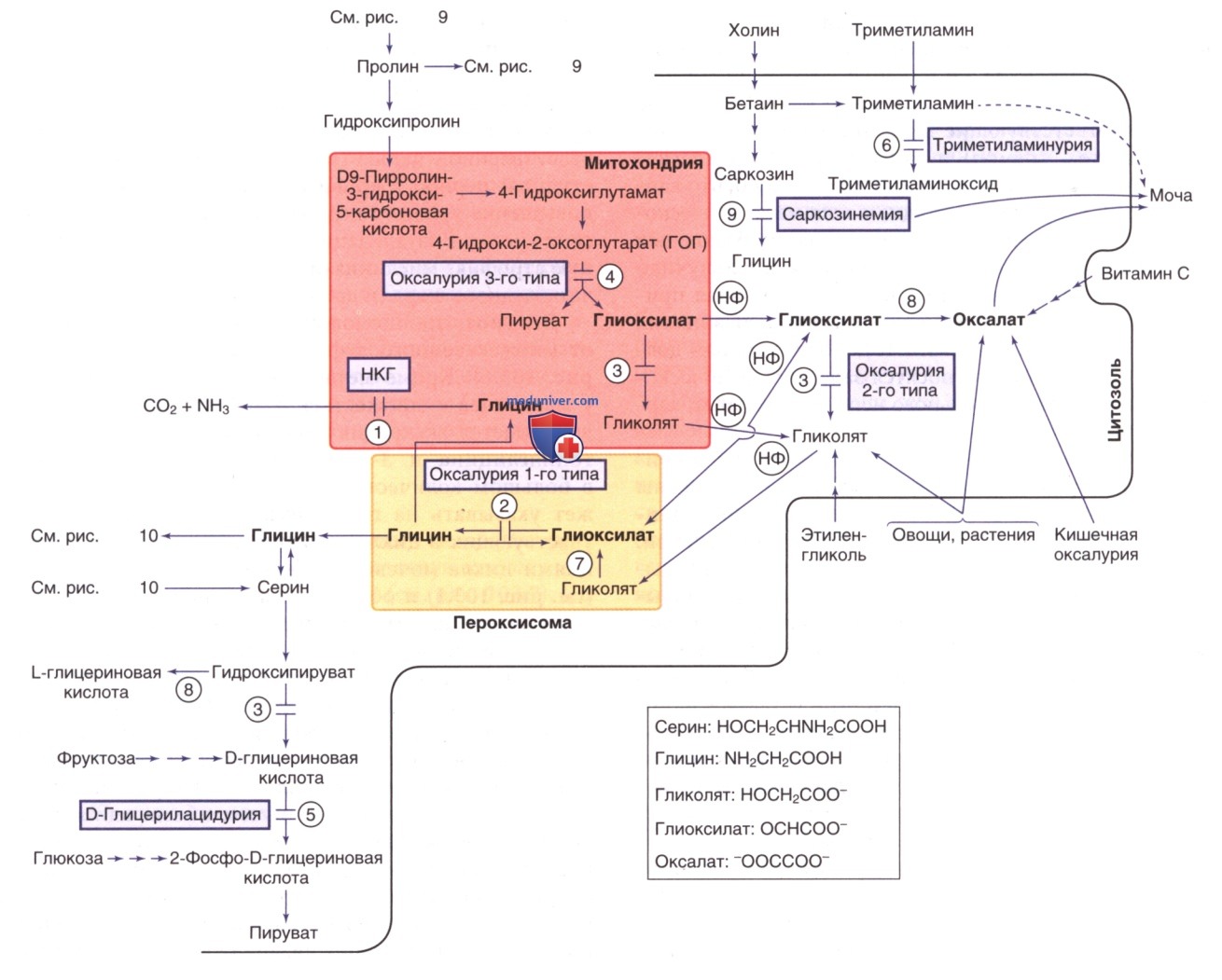
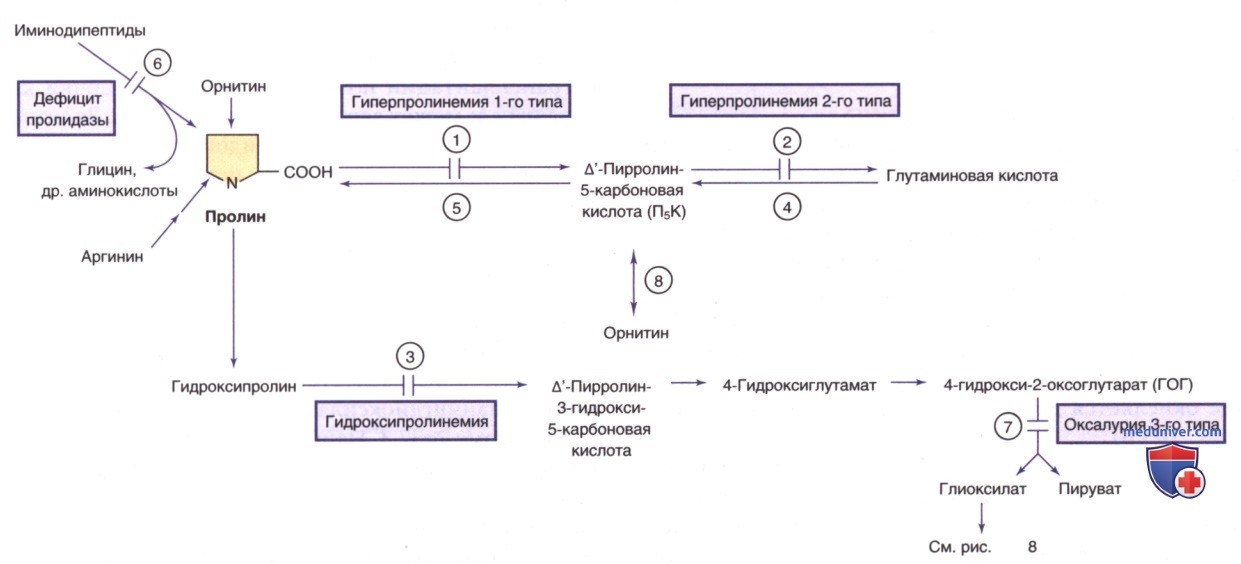
Пациенты поддаются лечению серином и глицином внутрь в случае, если оно началось в очень раннем возрасте. Рис. 8 и 10 демонстрируют метаболический путь синтеза и катаболизма серина.
а) Дефицит 3-фосфоглицератдегидрогеназы. Дефицит 3-фосфоглицератдегидрогеназы имеет широкий спектр симптомов и проявляется в разл. возрасте. Синдром Ноя-Лаксовой 1-го типа — наиболее тяжелая патология, которая в пренатальном периоде проявляется ЗВУР и ВПР, в т.ч. дисморфическими чертами лица, микроцефалией, ВПР ЦНС, деформацией конечностей и ихтиозом. Большинство пациентов с этой формой рождаются мертвыми/умирают в раннем неонатальном периоде.
Дефицит 3-фосфоглицератдегидрогеназы в младенчестве проявляется проблемами с кормлением, задержкой физического развития, рвотой, повышенной возбудимостью, судорогами, серьезной задержкой психического развития и гипертонусом, прогрессирующим в спастическую квадриплегию. У некоторых младенцев с такой патологией наблюдаются нистагм, катаракта, гипогонадизм и мегалобластная анемия.
У пациентов с более легкой формой этого нарушения имеются когнитивные нарушения, поведенческие проблемы, нейросенсорная полиневропатия и судороги, возникающие в раннем возрасте.
К лабораторным признакам относятся низкие уровни серина и глицина в плазме крови, взятой натощак, а также очень низкие уровни серина и глицина в СМЖ. В моче отсутствуют аномальные метаболиты органических кислот. На МРТ ГМ можно видеть церебральную атрофию с увеличенными желудочками, значительное уменьшение белого в-ва и нарушение миелинизации. Диагноз подтверждается анализом ДНК/измерением активности фермента в культуре фибробластов.
Лечение высокими дозами серина (внутрь 200-700 мг/кг в сутки) и глицина (200-300 мг/кг в сутки) нормализует содержание серина в крови и СМЖ. Если лечение начинается в постнатальном периоде, оно позволяет уменьшить судороги, спастичность и улучшить миелинизацию ГМ. Зарегистрирован один клинический случай, продемонстрировавший возможность предотвращения задержки психического развития в случае, если лечение начато в первые дни жизни/пренатальном периоде.
Заболевание наследуется по АуР-типу. Ген фермента 3-фосфоглицератдегидрогеназы (PHGDH) расположен в хромосоме 1p12. Если известны семейные патогенные варианты, то возможна молекулярная пренатальная диагностика. Прием серина матерью, вынашивающей плод с этой патологией, направлен на стабилизацию окружности головы плода, о чем свидетельствует УЗИ. Дополнительный прием серина продолжается в качестве послеродового лечения; к 4 годам такие пациенты неврологически оказались здоровы.
Относительно простое лечение приводит к «+» результату, поэтому очень важно диагностировать это заболевание у любого ребенка с микроцефалией и неврологическими нарушениями, напр. с задержкой психомоторного развития/судорожными расстройствами. Измерения серина и глицина в СМЖ имеют решающее значение для постановки диагноза, поскольку небольшое снижение указанных аминокислот в плазме может остаться незамеченным.
б) Дефицит фосфосеринаминотрансферазы. Фосфосеринаминотрансфераза-1 катализирует превращение 3-фосфогидроксипирувата в 3-фосфосерин (см. рис. 10). Дефицит этого фермента может проявляться в неонатальном периоде в виде проблем с питанием, цианотических приступов и повышенной возбудимости и может прогрессировать до трудноизлечимых многоочаговых судорог и микроцефалии. Визуализирующие исследования ГМ позволяют выявить генерализованную церебральную атрофию и атрофию мозжечка.
Лабораторные исследования плазмы крови, взятой после приема пищи, позволяют выявить нормальное/слегка пониженное содержание серина и глицина. При аминокислотном анализе СМЖ концентрации серина и глицина обычно понижены. Лечение серином и глицином, как указывалось ранее, может привести к клиническому улучшению. Состояние наследуется как АуР-признак, и ген (PSAT1) расположен в хромосоме 9q21.2.
в) Дефицит 3-фосфосеринфосфатазы. 3-Фосфосеринфосфатаза катализирует заключительную стадию синтеза L-серина, превращая 3-фосфосерин в L-серин. Дефицит этого фермента приводит к редкому нарушению с клиническими и биохим. признаками, неотличимыми от дефицита 3-фосфоглицератдегидрогеназы и фосфосеринаминотрансферазы-1. Заболевание вызвано АуР-патогенными вариантами PSPH, расположенного в хромосоме 7p11.2.