Нейромедиаторы представляют собой хим. в-ва, высвобождаемые из аксонов возбужденных нейронов в синаптических соединениях; они опосредуют инициацию и усиление/ингибирование нервных импульсов. Основную часть нейромедиаторов составляет ряд аминокислот и их метаболитов. Патогенные варианты генов, ответственных за синтез, транспорт и деактивацию этих в-в, могут приводить к состояниям, проявляющимся в виде неврологических и/или психических отклонений (табл. 3).
Раньше детям с патологией нейромедиаторов ставили синдромальные диагнозы, напр. церебральный паралич, эпилепсия, паркинсонизм, дистония и РАС. Диагностика в большинстве случаев требует проведения специализированных лабораторных исследований СМЖ, потому что некоторые из нейромедиаторов (дофамин и серотонин), вырабатываемых в ЦНС, не проникают через ГЭБ и их патологические концентрации не обнаруживаются в сыворотке крови/моче. В настоящее время выявляется все большее число таких состояний; заболевания, которые раньше считались редкими, теперь диагностируются все чаще.
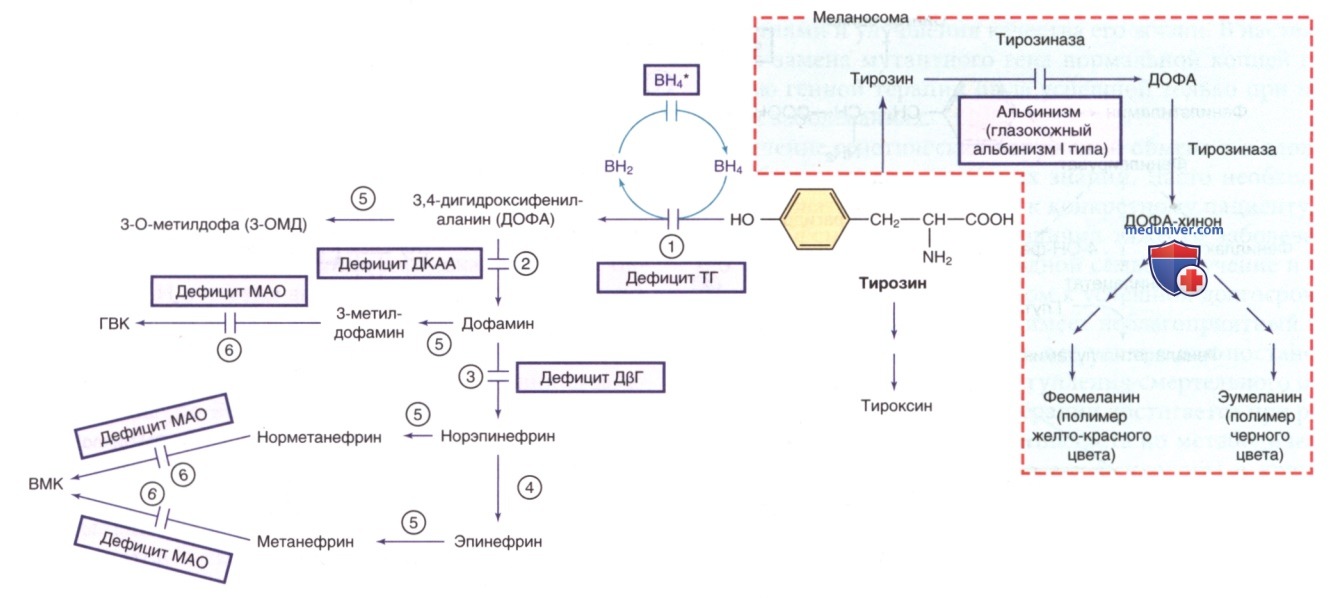
а) Дефицит тирозингидроксилазы (младенческий паркинсонизм, аутосомно-рецессивная дофамин-зависимая дистония, аутосомно-рецессивный синдром Сегава (Segawa)). Тирозингидроксилаза катализирует образование L-ДОФА из тирозина. Дефицит этого фермента приводит к дефициту дофамина и норэпинефрина (см. рис. 2). ДД включает широкий спектр наследственных дистоний, в т.ч. АуД-дистонию, вызванную дефицитом ГТФ-циклогидролазы.
Рисунок 2. Другие пути метаболизма тирозина. ВН4* указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). ГВК — го-мованиловая кислота; ВМК — ванилилминдальная кислота; ВН4 — тетрагидробиоптерин. Ферменты: 1) тирозингидроксилаза (ТГ); 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) дофамин-β-гидроксилаза (ДβГ); 4) фенилэтаноламин-N-метилтрансфераза (ФНМТ); 5) катехол-О-метилтрансфераза (КОМТ); 6) МАО
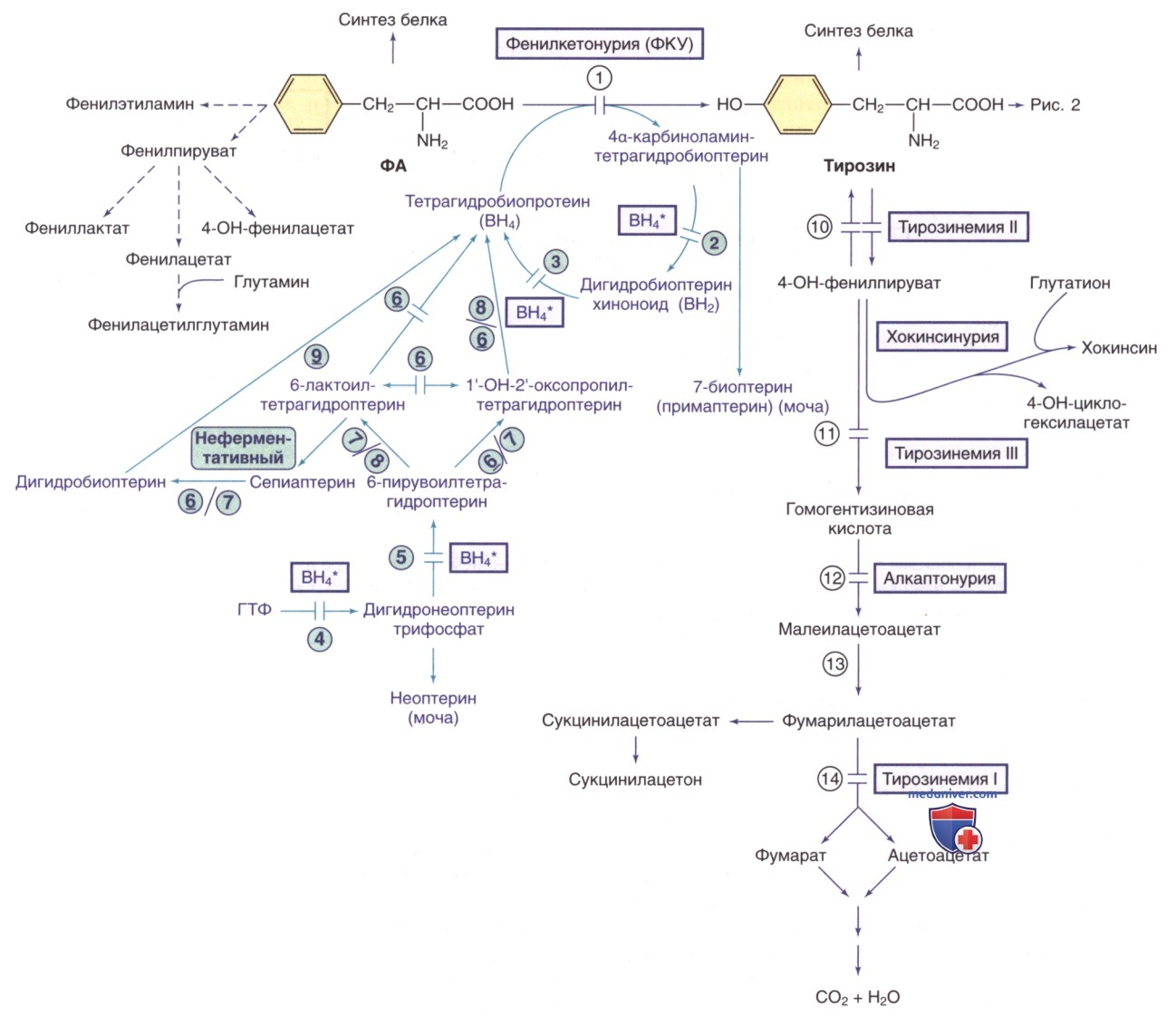
Рисунок 1. Пути метаболизма фенилаланина (ФА) и тирозина. Генетически детерминированные ферментативные нарушения показаны горизонтальными линиями, пересекающими стрелку(-и) реакции. Пути синтеза кофактора тетрагидробиоптерина (ВН4) показаны фиолетовым цветом. ВН4* относится к дефектам метаболизма ВН4, влияющим на гидроксилазы ФА, тирозина и триптофана (рис. 2 и 5). Ферменты: 1) фенилаланингидроксилаза; 2) птеринкарбиноламиндегидратаза; 3) дигидробиоптеринредуктаза; 4) гуанозинтрифосфат (ГТФ)-циклогидролаза; 5) 6-пирувоилтетрагидроптеринсинтаза; 6) сепиаптеринредуктаза; 7) карбонилредуктаза; 8) альдолазоредуктаза; 9) дигидрофолатредуктаза; 10) тирозинаминотрансфераза; 11) 4-гидроксифенилпируватдиоксигеназа; 12) диоксигеназа гомогентизиновой кислоты; 13) малеилацетоацетат изомераза; 14) фумарилацетоацетат гидролаза
Клинические проявления варьируют от легких до очень тяжелых. В целом было признано два фенотипа. В легкой форме (дофамин-зависимая дистония/тип А) возникают симптомы односторонней дистонии конечностей, вызывающие нарушение координации походки и постуральный тремор, которые появляются в детстве и прогрессируют с возрастом при отсутствии лечения. Могут наблюдаться суточные колебания симптомов (ухудшение в конце дня и улучшение после ночного сна). Когнитивные функции обычно в норме.
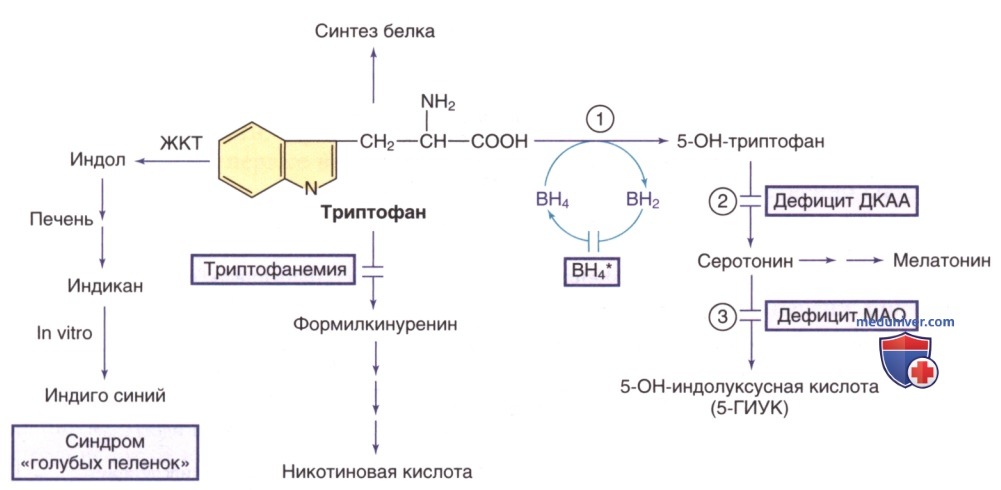
Рисунок 5. Пути метаболизма триптофана. Тетрагидробиоптерин (ВН4*) указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). Ферменты: 1) триптофангидроксилаза; 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) МАО
При тяжелой форме дефицита тирозингидроксилазы (младенческий паркинсонизм, детская энцефалопатия/тип В) клинические проявления возникают при рождении/вскоре после него и включают микроцефалию, задержку психического развития, непроизвольные движения конечностей со спастичностью, дистонию, птоз, амимичное лицо-маску, окулогирные кризы (движение глаз вверх) и вегетативную дисфункцию (нестабильность ТТ, повышенное потоотделение, гипогликемия, слюноотделение, тремор, желудочно-кишечный рефлюкс, запор). Могут отмечаться гиперрефлексия, миоклонус, атетоз и дистальная хорея. Пациенты с тяжелой формой обычно не в полной мере поддаются лечению L-ДОФА и склонны к развитию такого побочного эффекта, как L-ДОФА -индуцированная дискинезия.
Реже при тяжелом течении энцефалопатии в первом полугодии жизни м.б. проявления выраженной гипокинезии и прогрессирующей гипотонии туловища в сочетании с фокальной/генерализованной дистонией, иногда с дистоническими кризами в течение нескольких дней и чрезмерными отрывистыми движениями (миоклонус и тремор без суточных колебаний)1.
К лабораторным признакам относятся пониженное содержание дофамина и его метаболита гомованилиновой кислоты и нормальные концентрации ВН4, неоптерина и 5-гидроксииндолуксусной кислоты (метаболит серотонина) в СМЖ.
Концентрация гомованилиновой кислоты и ее отношение к 5-гидроксииндолуксусной кислоте в СМЖ коррелируют с возрастом начала заболевания и тяжестью фенотипа.
Содержание пролактина в сыворотке крови обычно повышено. Эти проявления не относятся к ДК заболевания; диагноз должен быть установлен на основании данных молекулярно-генетического исследования.
Лечение L-ДОФА/карбидопой приводит к значительному клиническому улучшению у большинства пациентов, но тяжелые формы неизменно сопровождаются L-ДОФА-индуцированными дискинезиями. Чтобы свести к минимуму побочные эффекты терапии, лечение следует начинать с низкой дозы и при необходимости увеличивать ее очень медленно. Др. терапевтические вмешательства включают антихолинергические ЛП, серотонинергические агенты и ингибиторы МАОВ, в т.ч. амантадин, бипериден и селегилин. В одном случае клинически эффективной оказалась билатеральная глубокая стимуляция субталамического ядра ГМ. Дефицит тирозингидроксилазы наследуется как АуР-признак. Молекулярное исследование патогенных вариантов в гене TH доступно в клинической практике.
б) Дефицит декарборксилазы ароматических L-аминокислот. Декарбоксилаза ароматических L-аминокислот представляет собой витамин-В6-зависимый фермент, катализирующий декарбоксилирование 5-гидрокситриптофана с образованием серотонина (см. рис. 5), и L-ДОФА с образованием дофамина (см. рис. 2). Клинические проявления обусловлены уменьшением доступного количества дофамина и серотонина. У новорожденных с этой патологией наблюдались нарушения глотания, вялость, гипотония, гипотермия, окулогирные кризы и птоз век. Клинические признаки у грудных детей и детей старшего возраста включают задержку психического развития, гипотонию туловища с гипертонусом конечностей, окулогирные кризы, экстрапирамидные двигательные расстройства (хореоатетоз, дистонию, миоклонус), нарушения ВНС (потливость, заложенность носа, слюноотделение, повышенную возбудимость, нестабильность ТТ, артериальную гипотензию). Симптомы могут меняться в течение дня, усиливаясь к вечеру.
К лабораторным признакам относится снижение концентрации дофамина и серотонина и их метаболитов (гомованилиновой кислоты, 5-5-гидроксииндолуксусной кислоты, норэпинефрина, ванилилминдальной кислоты) и повышение уровня 5-гидрокситриптофана, L-ДОФА и его метаболита (3-О-метилдофа) в физиол. жидкостях, особенно в СМЖ. Также наблюдались повышенные концентрации пролактина в сыворотке (в результате дефицита дофамина). МРТ ГМ выявляет церебральную атрофию с дегенеративными изменениями белого в-ва. Программа скрининга мочи с определением 3-О-метилдофа и ваниллилминдальной кислоты оказалась диагностически перспективной в группах населения с высокой частотой встречаемости заболевания.
Лечение предшественниками нейромедиаторов дало временное клиническое улучшение. Допамин («Дофамин») и серотонин не имеют терапевтической значимости, поскольку они неспособны преодолевать ГЭБ. Были испытаны агонисты дофамина (L-ДОФА/карбидопа, бромокриптин), ингибиторы МАО (транилципромин), серотонинергические агенты и высокие дозы пиридоксина, кофактора фермента декарбоксилазы ароматических L-аминокислот. Прием БАД пиридоксина может оказать «+» влияние на пациентов с вариантом p.S250F в декарбоксилазе ароматических L-аминокислот. Недавние исследования ЦНС-направленной генной терапии с применением аденовирусного вектора показали эффективность у некоторых пациентов.
В тайваньской популяции с высокой частотой встречаемости заболевания была достигнута преимплантационная генетическая диагностика после экстакорпорального оплодотворения. Ген, кодирующий декарбоксилазу ароматических L-аминокислот (DDC), расположен в хромосоме 7p12.1. Заболевание наследуется по АуР-типу.
в) Дефицит тетрагидробиоптерина. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше. ВН4 является кофактором ФАГ (см. рис. 1), тирозингидроксилазы (см. рис. 2), триптофангидроксилазы (см. рис. 5) и синтазы оксида азота. Его синтез из ГТФ осуществляется во многих тканях (см. рис. 1). Дефицит ферментов, участвующих в биосинтезе ВН4, приводит к недостаточному образованию этого кофактора, что вызывает дефицит моноаминных нейромедиаторов с/без сопутствующей ГФА.
1. Дефицит тетрагидробиоптерина с гиперфенилаланинемией. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
2. Дефицит тетрагидробиоптерина без гиперфенилаланинемии:
- Дефицит ГТФ-циклогидролазы-1 (наследственная прогрессирующая дистония, аутосомнодоминантная дофа-чувствительная дистония, аутосомно-доминантный синдром Сегава). Эта форма дистонии, вызванная дефицитом ГТФ-циклогидролазы-1, наследуется по АуД-типу и чаще встречается у женщин, чем у мужчин (Ж/М 4:1). Клинические проявления обычно начинаются в раннем детстве с тремора и дистонии нижних конечностей (походка на пальцах), которые в течение нескольких лет могут распространиться на кисти и стопы. Дистонии нижних конечностей могут предшествовать тортиколлис (спастическая кривошея), дистония верхних конечностей и нарушение координации. Развитие в раннем возрасте в целом соответствует норме. Степень выраженности симптомов заметно изменяется в течение суток, усиливаясь к вечеру и уменьшаясь после сна. Нередко встречается вегетативная лабильность. С возрастом также может присут-ствовать/развиваться паркинсонизм.
Зарегистрированы случаи манифестации заболевания в позднем взрослом возрасте с дистонии действия («писчий спазм»), торти-коллиса и генерализованного ригидного гипертонуса с тремором, но без постуральной дистонии. Кроме того, по имеющимся ограниченным данным о взрослых предполагается наличие симптомов, обусловленных с дефицитом серотонина (нарушение сна, когнитивные нарушения, импульсивность).
Выделяют два типа заболевания: классическую постуральную дистонию (нарушение тонуса мышц при поддержании позы) и фазическую дистонию (сочетание дистонии при движениях и постуральной дистонии).
К лабораторным признакам относится снижение уровня BH4 и неоптерина в СМЖ при отсутствии ГФА. Содержание дофамина и его метаболита (гомованилиновой кислоты) в СМЖ м.б. понижено. Серотонинергический путь в меньшей степени подвержен дефициту этого фермента, и поэтому концентрации серотонина и его метаболитов обычно соответствуют норме. Содержание ФА в плазме в норме, но нагрузочный пероральный тест с фенилаланином (100 мг/кг) показывает аномально высокий уровень ФА в плазме с повышенным соотношением ФА/тирозин. Соотношение, полученное через 2-3 ч после нагрузки, в комбинации с уровнем неоптерина в моче обладает оптимальной диагностической ЧС. Возможность бессимптомного носительства указывает на то, что в патогенезе могут играть роль и др. факторы/гены. Бессимптомный носитель м.б. выявлен при проведении нагрузочного теста с фенилаланином.
Диагноз подтверждают определением пониженного содержания BH4 и неоптерина в СМЖ, измерением активности фермента и молекулярно-генетическим анализом. Клинически следует проводить ДД этого заболевания от др. причин дистонии и детского паркинсонизма, особенно от дефицитов тирозингидроксилазы, сепиаптеринредуктазы и декарбоксилазы ароматических аминокислот.
Лечение L-ДОФА/карбидопой обычно позволяет достичь резкого клинического улучшения. Прием BH4 внутрь также эффективен, но применяется редко.
3. Дефицит сепиаптеринредуктазы. Сепиаптеринредуктаза участвует в превращении 6-пирувоилтетрагидроптерина в BH4. Она также принимает участие в реутилизационном пути синтеза BH4 (см. рис. 1). Дефицит сепиаптеринредуктазы приводит к накоплению 6-лактоилтетрагидроптерина, который м.б. преобразован в сепиаптерин неферментативным путем. Большая часть сепиаптерина метаболизируется до ВН4 посредством реутилизационного пути в периферических тканях (см. рис. 1), но из-за низкой активности дигидрофолатредуктазы в ГМ количество BH4 остается недостаточным для надлежащего синтеза дофамина и серотонина. Этим объясняется отсутствие ГФА и часто отсроченная диагностика.
Клинические проявления обычно развиваются в первые месяцы жизни. Кардинальные проявления включают приступообразную скованность, окулогирные кризы и гипотонию. К дополнительным признакам относятся задержка в развитии моторики и речи, слабость, гипертонус конечностей, дистония, гиперрефлексия и паркинсонизм с ранним началом. Симптомы обычно меняются в течение суток. Зачастую ошибочно диагностируют ДЦП, при этом зарегистрирован широкий спектр симптомов. Диагноз устанавливают путем измерения нейромедиаторов и метаболитов птерина в СМЖ, которые выявляют пониженные концентрации дофамина, гомованилиновой кислоты, норэпинефрина и 5-гидроксииндолуксусной кислоты и заметно повышенное содержание сепиаптерина и дигидробиоптерина. Концентрация пролактина в сыворотке м.б. повышена. Нагрузочный тест с фенилаланином м.б. диагностически значимым.
Резкое клиническое улучшение обычно наступает при лечении медленно увеличиваемыми дозами L-ДОФА/карбидопы и 5-гидрокситриптрфана. Заболевание наследуется по АуР-типу; ген SPR, кодирующий сепиаптеринредуктазу, расположен в хромосоме 2р13.2.
г) Дефицит дофамин-бета-гидроксилазы. Дофамин-β-гидроксилаза катализирует превращение дофамина в норэпинефрин (см. рис. 2). Дефицит этого фермента приводит к снижению/отсутствию синтезируемого норэпинефрина, что в свою очередь приводит к нарушению регуляции симпатической функции. Дети грудного и старшего возраста могут испытывать трудности при открывании глаз, птоз, гипотонию, гипотермию, гипогликемию и заложенность носа. У взрослых пациентов может наблюдаться глубокий дефицит регуляции ВНС, приводящий к тяжелой ортостатической гипотензии и сексуальной дисфункции у мужчин. Предобморочная симптоматика включает головокружение, нечеткость зрения, одышку, дискомфорт в области шеи и боль в груди; обоняние остается относительно сохранным.
Для диагностики м.б. полезным тестирование функции ВНС (измерение коэффициента синусовой аритмии, исследование АД во время контролируемой гипервентиляции, проба Вальсальвы (Valsalva), холодовый прессорный тест, упражнение на сжатие кистей рук). К лабораторным признакам относится снижение/отсутствие норэпинефрина и адреналина и их метаболитов при повышенном уровне дофамина и его метаболита (гомованилиновой кислоты) в плазме, СМЖ и моче. Патогномоничным признаком этого заболевания м.б. повышенный уровень дофамина в плазме. На МРТ ГМ заметно уменьшение объема ГМ, что согласуется с нейротрофической ролью норадреналина. Лечение L-дигидроксифенилсерином, который превращается в норэпинефрин непосредственно in vivo под действием декарбоксилазы ароматических L-аминокислот, приводит к значительному уменьшению ортостатической гипотензии и нормализует содержание норадреналина и его метаболитов.
Состояние наследуется по АуР-типу; ген (DBH), кодирующий дофамин-β-гидроксилазу, расположен в хромосоме 9q34.2.
д) Дефицит моноаминоксидазы А (МАО-А). Геном человека кодирует два изофермента МАО: МАО-А и МАО-В. Оба фермента катализируют окислительное дезаминирование большинства биогенных аминов в организме, в т.ч. серотонина (см. рис. 5), норадреналина, адреналина и дофамина (см. рис. 2). Гены обоих изоферментов расположены в Х-хромосоме (Xp11.З), находясь в непосредственной близости. Делеция обоих генов может захватывать соседний ген NDP, вызывая синдром смежной делеции, который проявляется атипичной болезнью Норри (Norrie). У пациентов мужского пола с дефицитом МАО-А наблюдается пограничный дефицит интеллектуального развития и сниженный контроль импульсов. Последствия изолированного дефицита МАО-В до конца не изучены. Комбинированный дефицит МАО-А и МАО-В вызывает тяжелую умственную отсталость и поведенческие проблемы. Он может сопровождаться выраженными лабораторными изменениями [напр., 4-6-кратное повышение серотонина в физиол. жидкостях, повышенное содержание метаболитов О-метилированного амина и пониженное содержание продуктов дезаминирования (ванилилминдальной кислоты, гомованилиновой кислоты)].
Коррекция режима питания (низкое потребление тирамина, фенилэтиламина и L-ДОФА/дофамина) не нормализовала уровень серотонина в крови пациентов. Дефицит МАО наследуется по Х-сцепленному типу. Лечение дефицита МАО-А поддерживающее.
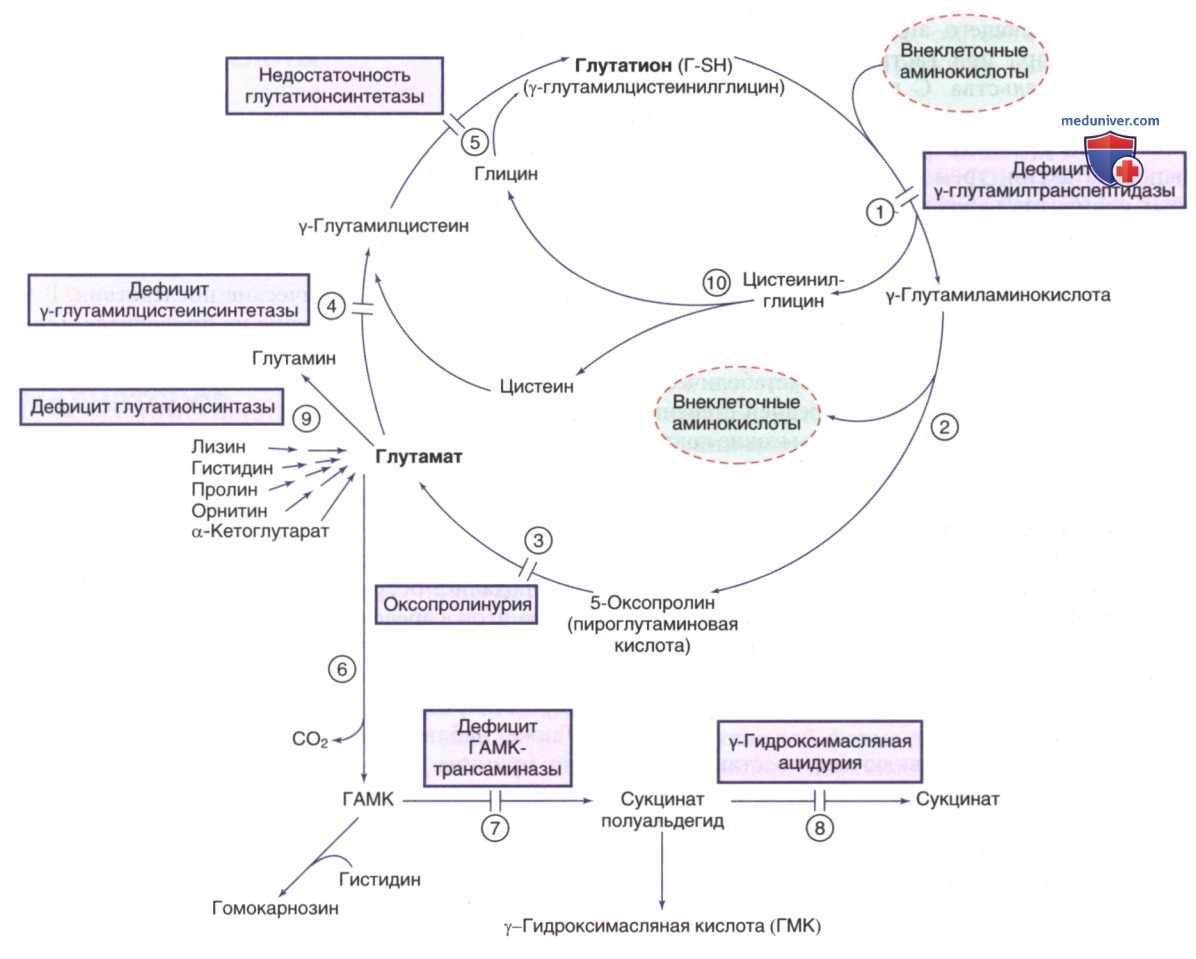
е) Нарушение метаболизма γ-аминомасляной кислоты (ГАМК). ГАМК является основным ингибиторным нейромедиатором, синтезируемым в синапсах при декарбоксилировании глутаминовой кислоты глутаматдекарбоксилазой (ГДКА). Этот же путь отвечает за продуцирование ГАМК в др. органах, особенно в почках и β-клетках ПЖЖ. Для фермента ГДКА требуется пиридоксин (витамин В6) в качестве кофактора. Идентифицированы два фермента ГДКА: ГДКА1 (ГДКА67) и ГДКА2 (ГДКА65). ГДКА1 является главным ферментом ГМ, а ГДКА2 представляет собой главный фермент β-клеток. АТл к ГДКА65 и ГДКА67 участвуют в развитии СД-1 и синдрома мышечной скованности (синдром ригидного человека) соответственно. ГАМК катаболизируется до янтарной кислоты двумя ферментами: ГАМК-трансаминазой и сукцинатполуальдегиддегидрогеназой (см. рис. 11).
Рисунок 11. γ-Глутамиловый цикл и связанные с ним пути. Отмечены нарушения синтеза и распада глутатиона (Г-SH). Ферменты: 1) γ-глутамилтранспептидаза (ГГТ); 2) у-глутамилциклотрансфераза; 3) 5-оксопролиназа; 4) у-глутамилцистеинсинтетаза; 5) глутатионсинтетаза; 6) декарбоксилаза глутаминовой кислоты; 7) трансаминаза гамма-аминомасляной кислоты (ГАМК); 8) сукцинат-полуальдегиддегидрогеназа; 9) глутаминсинтетаза; 10) дипептидаза
1. Дефицит трансаминазы γ-аминомасляной кислоты. Клинические проявления у двух сиблингов грудного возраста с подтвержденным индексом отцовства включали выраженную задержку психомоторного развития, гипотонию, гиперрефлексию, летаргию, рефрактерные судороги и повышенный линейный рост, вероятно, обусловленный ГАМК-опосредованной повышенной секрецией гормона роста. В СМЖ были обнаружены повышенные концентрации ГАМК и β-аланина (см. рис. 11). При патологоанат. исследовании ГМ были отмечены признаки лейкодистрофии. У третьего пациента наблюдались тяжелое психомоторное отставание в развитии, рецидивирующая эпизодическая летаргия и трудноизлечимые приступы с нарушениями метаболитов СМЖ, сравнимые с аналогичными нарушениями у исследуемых пробандов. Дефицит трансаминазы ГАМК выявляется в клетках ГМ и лимфоцитах.
Лечение симптоматическое. Терапия витамином В6 (кофактором фермента) оказалась неэффективной. Ген (АВАТ) расположен в хромосоме 16р13.2; заболевание наследуется по АуР-типу.
2. Дефицит сукцинатполуальдегиддегидрогеназы (γ-гидроксимасляная ацидурия). Клинические проявления дефицита сукцинатполуальдегиддегидрогеназы обычно начинаются в младенчестве с задержки психического развития с непропорциональным дефицитом экспрессивной речи, гипотонией и атаксией; судороги отмечаются у 50% пациентов (см. рис. 11). Многим пациентам диагностируют РАС. Нейропсихиатрическая сопутствующая патология (особенно оппозиционно-вызывающее расстройство поведения, навязчивые идеи и гиперактивность) может приводить к инвалидности, особенно у подростков и взрослых. К аномальным признакам на ЭЭГ относятся фоновое замедление и генерализованные спайк-волновые пароксизмы, с переменной латерализацией по полушариям и преобладанием напряжения. Сообщалось о фоточувствительности и электрографическом эпилептическом статусе во сне в сочетании с трудностями в поддержании сна и чрезмерной дневной сонливостью.
На МРТ ГМ можно увидеть усиление на Т2-взвешенном гиперинтенсивном изображении, затрагивающее бледный шар, зубчатые ядра мозжечка и субталамические ядра, как правило, с двусторонним симметричным распределением.
Биохим. маркером является повышение γ-гидроксимасляной кислоты в физиол. жидкостях (СМЖ, плазме, моче) у всех пациентов. В СМЖ обнаруживаются повышенные концентрации ГАМК. Повышенный уровень γ-гидроксимасляной кислоты в моче позволяет предположить данное заболевание, диагноз верифицируется посредством молекулярно-генетического тестирования.
Лечение остается неясным; вигабатрин (ингибитор ГАМК-трансаминазы) использовался эмпирически с неоднозначными результатами, и его применение вызывает опасения, поскольку он дополнительно повышает уровень ГАМК в ЦНС при заболевании, которое уже сопровождается увеличением концентрации ГАМК. Кроме того, вигабатрин может вызывать сужение полей зрения, поэтому его длительное применение противопоказано.
Ген сукцинатполуальдегиддегидрогеназы (ALDH5A1) расположен в хромосоме 6р22, наследование происходит по АуР-типу. Пренатальную диагностику проводили путем измерения уровня у-гидроксимасляной кислоты в околоплодных водах, анализа активности ферментов в амниоцитах, взятия проб ворсинок хориона и ДНК-анализа.
ж) Дефекты нейромедиаторных транспортных белков. В транспорте разл. нейромедиаторов через мембраны нейронов задействованы >20 белков. Основной функцией большинства этих транспортных белков является удаление избыточного количества нейромедиаторов из синапса обратно в пресинаптические нейроны (обратный захват). Этот процесс рециркуляции не только регулирует точное действие нейромедиаторов в синапсе, но и возвращает пресинаптическим нейронам нейромедиаторы для последующего использования. Некоторые транспортные белки задействованы в переносе нейромедиаторов из цитоплазмы нейронов через мембрану синаптических везикул для хранения (везикулярные транспортеры). При стимуляции нейронов эти везикулы посредством экзоцитоза высвобождают квант1 нейромедиатора. Патогенные варианты транспортных белков ожидаемо препятствуют правильному обратному захвату и хранению нейромедиаторов и могут приводить к клиническим проявлениям, аналогичным тем, которые наблюдаются при нарушениях метаболизма нейромедиаторов.
Описано несколько заболеваний, вызванных патогенными вариантами нейромедиаторных транспортных белков, в т.ч. дефицит транспортного белка дофамина и болезнь везикулярного транспортера дофамина-серотонина.
1. Дефицит транспортного белка дофамина. Этот белок-транспортер участвует в обратном захвате дофамина пресинаптическими нейронами; его дефицит приводит к истощению дофамина и, следовательно, к состоянию дефицита дофамина. Белок-транспортер дофамина кодируется геном SLC6A3, локализованным на хромосоме 5р15.33. Зарегистрированы 13 детей с патогенными вариантами этого гена. У них отмечались проявления синдрома инфантильной паркинсонизм-дистонии. Вскоре после рождения развивались повышенная возбудимость и затруднения вскармливания, которые прогрессировали до гипотонии, паркинсонизма, дистонии, навык удержания головы не формировался, у детей имело место глобальное отставание в развитии в грудном возрасте.
В связи со схожестью клинических проявлений большое количество больных длительно наблюдаются и проходят лечение с ошибочным клиническим диагнозом ДЦП. МРТ ГМ обычно не выявляет отклонений.
Исследование СМЖ показало повышенное содержание γ-гидроксимасляной кислоты и нормальный уровень 5-гидроксииндолуксусной кислоты. Содержание γ-гидроксимасляной кислоты в моче и концентрация пролактина в сыворотке были повышены. Диагноз был установлен при обнаружении мутации с потерей функции в гене SLC6A3. Эффективного лечения не разработано; применение L-ДОФА/карбидопы не приводило к улучшению клинических/биохим. показателей.
2. Болезнь везикулярного транспортера дофамина-серотонина (дефицит везикулярного транспортера моноаминов). Это АуР-заболевание, описанное у восьми детей в семье с близкородственными браками из Саудовской Аравии, вызвано патогенным вариантом гена SLC18A2. Этот ген кодирует везикулярный транспортер моноаминов 2, который участвует в транспортировке дофамина и серотонина из цитоплазмы в синаптические везикулы для хранения, расположенные в пресинаптических терминалях аксонов пресинаптических нейронов. У большинства детей с этой патологией в первый год жизни наблюдались симптомы, соответствующие дефициту дофамина (гипотония, прогрессирующая до дистонии, паркинсонизм, окулогирные кризы), серотонина (нарушения сна и психические расстройства) и норадреналина-адреналина (повышенное потоотделение, тремор, нестабильность ТТ, постуральная гипотензия, птоз).
Задержка нейрокогнитивных функций выявляется также на первом году жизни. Суточной вариабельности симптомов не отмечалось. Результаты визуализирующих исследований ГМ были в пределах нормы. Изменения уровней нейромедиаторов ЦНС и их метаболитов были непостоянными.
Фенотип сходен с тем, который наблюдается при дефиците декарбоксилазы ароматических L-аминокислот и ВН4. Для диагностики необходимо проведение молекулярного анализа SLC18A2 (расположен в хромосоме 10q25.3). Лечение L-ДОФА/карбидопой приводило к обострению симптомов, а на фоне лечения прамипексолом (агонистом рецепторов дофамина) отмечались многообещающие клинические результаты.
з) Дефицит гистидиндекарбоксилазы. При декарбоксилировании гистидина гистидиндекарбоксилазой образуется гистамин, который выполняет функции нейромедиатора в ГМ. Дефицит этого фермента (в основном в заднем гипоталамусе) приводит к дефициту гистамина в ЦНС, а в одной семье пациентов вызвал АуД-форму синдрома Туретта (Tourette).
и) Гиперпролинемия. У большинства пациентов с гиперпролинемией типов I и II наблюдаются общие симптомы, такие как умственная отсталость и судороги. Гиперпролинемия типа I обычно характеризуется доброкачественным клиническим течением, но имеется повышенный риск развития шизофрении. Роль повышенной концентрации пролина в патогенезе шизофрении остается невыясненной. Неврологические нарушения, наблюдаемые при гиперпролинемии типа II, в основном обусловлены развитием зависимости от витамина В6 при этом заболевании. Коррекция режима питания при гиперпролинемии типов I и II нецелесообразна и не рекомендуется.
к) Дефицит 3-фосфоглицератдегидрогеназы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
л) Дефицит фосфосеринаминотрансферазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
м) Некетотическая гиперглицинемия. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.