Пролин является заменимой аминокислотой, эндогенно синтезируемой из глутаминовой кислоты, орнитина и аргинина (см. рис. 9). Пролин и гидроксипролин в высоких концентрациях содержатся в коллагене. Обычно ни одна из этих аминокислот не обнаруживается в моче в больших количествах. Экскреция пролина и гидроксипролина в виде иминопептидов (дипептидов и трипептидов, содержащих пролин/гидроксипролин) увеличивается при нарушениях ускоренного обмена коллагена, напр. при рахите/гиперпаратиреозе.
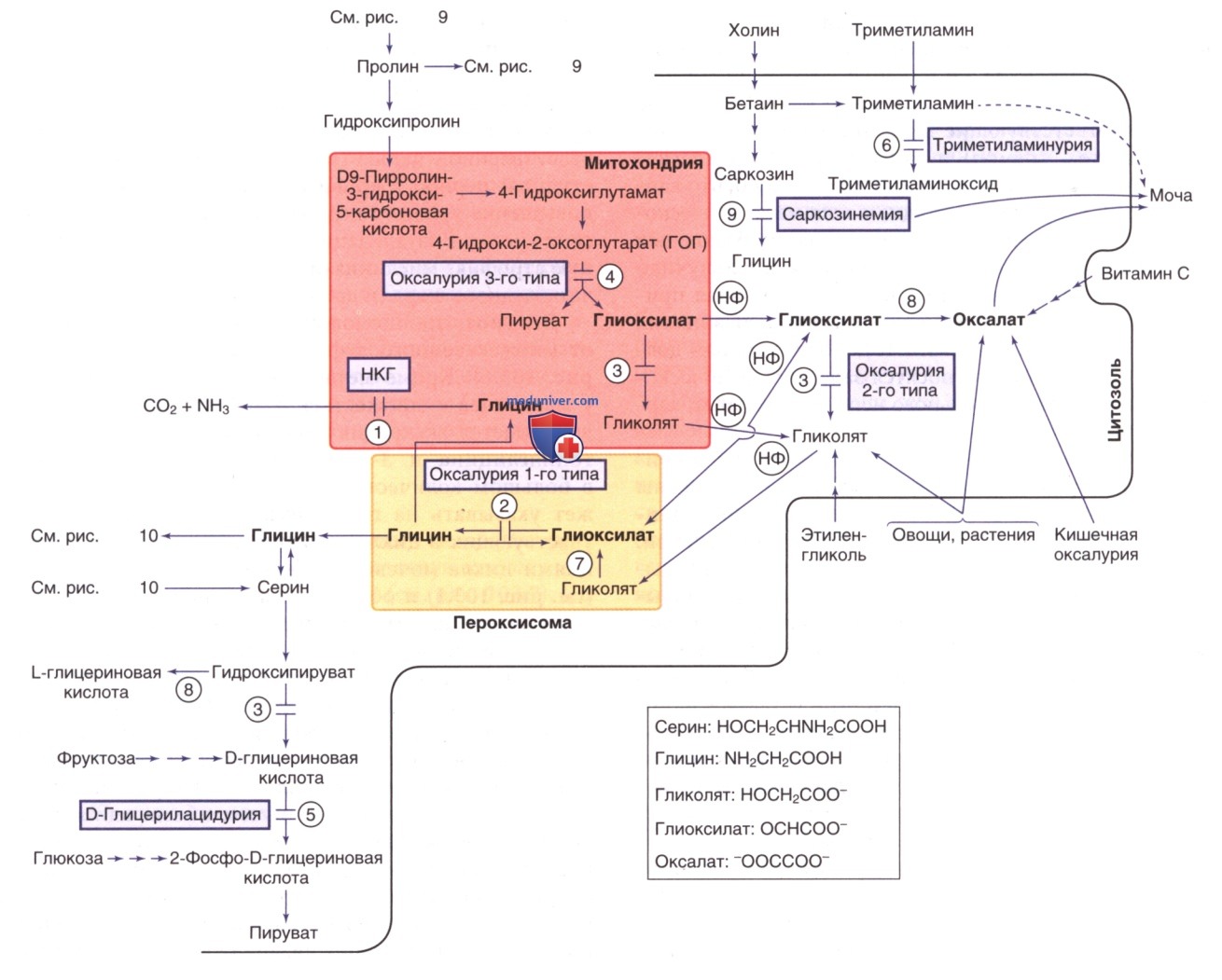
Пролин также содержится в синапсах, где он может взаимодействовать с рецепторами глицина и глутамата. Катаболический путь пролина и гидроксипролина продуцирует глиоксиловую кислоту, которая в дальнейшем может метаболизироваться до глицина/щавелевой кислоты (см. рис. 8).
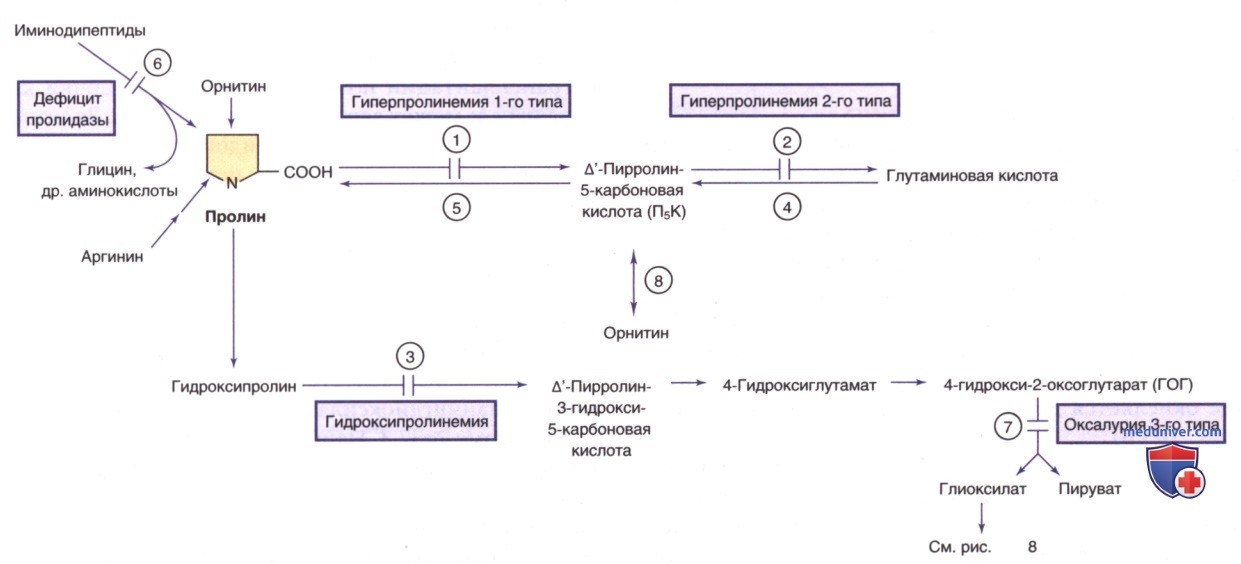
Гиперпролинемия 1-го и 2-го типа вызывает накопление пролина в тканях. Снижение синтеза пролина de novo вызывает такие синдромы, как халазодермия* с прогероидными признаками/спастической параплегией. Ниже описаны два типа первичной гиперпролинемии.
а) Гиперпролинемия типа I. Это редкое АуР-заболевание вызвано дефицитом пролиноксидазы (пролиндегидрогеназа; см. рис. 9). Большинство пациентов с гиперпролинемией 1-го типа не имеют клинических проявлений, хотя у некоторых может наблюдаться умственная отсталость, судороги и поведенческие проблемы. Гиперпролинемия также м.б. фактором риска РАС и шизофрении. Природа такого широкого фенотипического диапазона в этом биохим. состоянии не установлена.
Ген, кодирующий пролиноксидазу (PRODH), картирован в хромосоме 22q11.2 и расположен в критической области велокардиофациального синдрома. Лабораторные исследования выявляют высокие концентрации пролина в плазме, моче и СМЖ. Также наблюдается повышенная экскреция с мочой гидроксипролина и глицина, что м.б. обусловлено насыщением общего механизма канальцевой реабсорбции вследствие обширной пролинурии.
Эффективного лечения пока не разработано. Ограничение пролина в питании вызывает небольшое снижение пролина в плазме, но клиническая польза этого подхода не доказана.
б) Гиперпролинемия типа II. Гиперпролинемия типа II представляет собой редкое АуР-заболевание, вызванное дефицитом Δ1-пирролин-5-карбоксилатдегидрогеназы (альдегиддегидрогеназа-4; см. рис. 9). У детей с такой патологией наблюдались судороги и умственная отсталость (обычно вызванные интеркуррентной инфекцией), но также известны случаи бессимптомных пациентов.
Причина таких несопоставимых клинических результатов до конца не изучена. Ген, кодирующий Δ1-пирролин-5-карбоксилатдегидрогеназу (ALDH4A1), расположен в хромосоме 1p36.13.
Лабораторные исследования показывают повышенные концентрации пролина и Δ1-пирролин-5-карбоновой кислоты в крови, моче и СМЖ. Присутствие этой кислоты отличает это заболевание от гиперпролинемии типа I. Повышенный уровень Δ1-пирролин-5-карбоновой кислоты в физиол. жидкостях, особенно в ЦНС, по-видимому, вызывает антагонизм к витамину В6 и приводит к зависимости от витамина В6.
Эта зависимость, вероятно, является основной причиной судорог и неврологических проявлений заболевания, и, т.о., можно объяснить вариабельность клинических проявлений у разных пациентов. Рекомендуется лечение высокими дозами витамина В6.
в) Дефицит пролидазы. При разложении коллагена образуются имидодипептиды, которые обычно расщепляются пролидазой в ткани. Дефицит пролидазы, наследуемый как АуР-признак, приводит к накоплению имидодипептидов в физиол. жидкостях. Возраст проявления заболевания варьирует от 6 мес до 30 лет.
Клинические признаки этого редкого нарушения различны и включают рецидивирующие, тяжелые и болезненные язвы кожи, обычно возникающие на руках и ногах. На несколько лет язве могут предшествовать др. поражения кожи, напр. отрубевидная эритематозная макулопапулезная сыпь, геморрагическая сыпь и телеангиэктазия. Большинство язв инфицируются. Заживление может занять несколько месяцев.
К др. признакам относятся задержка психического развития, умственная отсталость, органомегалия, анемия, тромбоцитопения и дисфункция иммунной системы, что приводит к повышенной восприимчивости к инфекциям (рецидивирующий средний отит, синусит, респираторная инфекция, спленомегалия). У некоторых пациентов могут присутствовать черепно-лицевые аномалии, такие как птоз, проптоз глаза, гипертелоризм, небольшой клювовидный нос и выступающие черепные швы. Были зарегистрированы бессимптомные случаи.
У детей отмечается рост заболеваемости СКВ. ДК является высокий уровень экскреции имидодипептидов с мочой. Ген пролидазы (PEPD) расположен в хромосоме 19q13.ll. Диагноз подтверждается ДНК-анализом. Ферментный анализ м.б. выполнен на эритроцитах/культуре фибробластов кожи.
Лечение дефицита пролидазы поддерживающее. Инфекционные осложнения могут привести к летальному исходу и требуют тщательного и упреждающего лечения АБ. Прием внутрь пролина, аскорбиновой кислоты и марганца, а также местное применение пролина и глицина оказались эффективными не у всех пациентов.
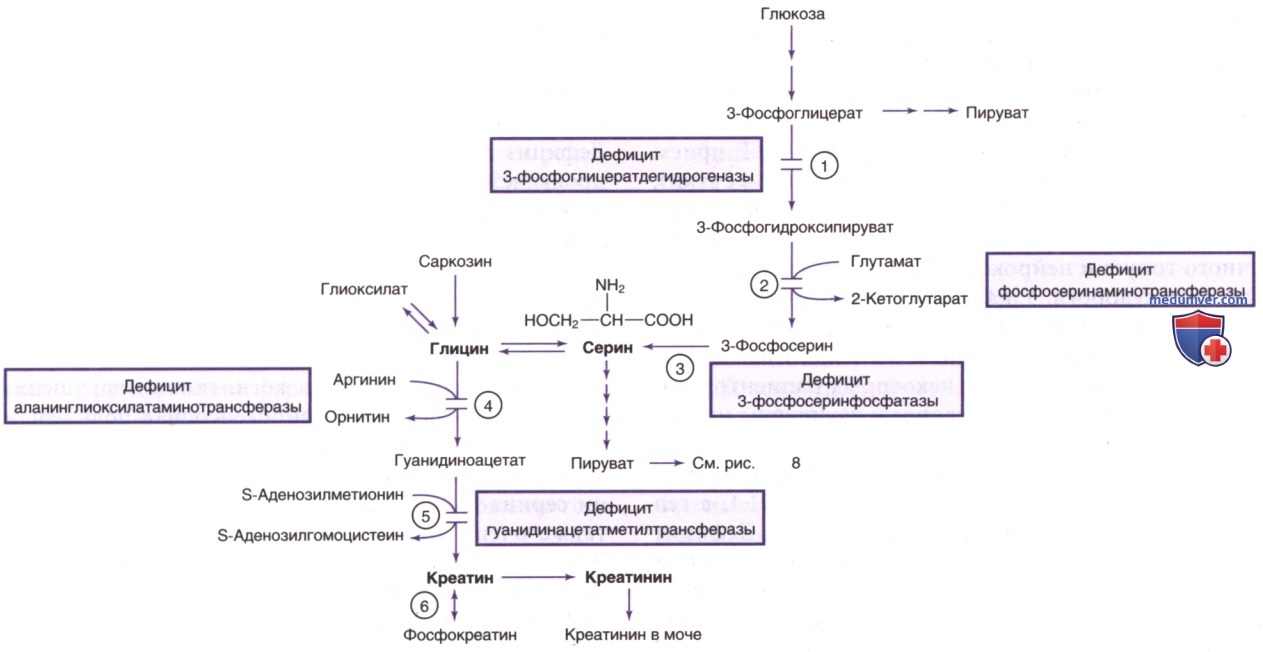
г) Нарушения синтеза пролина de novo. Синтез de novo пролина и орнитина из глутамата имеет важнейшее значение для нормальной биологии соединительной ткани и поддержания цикла мочевины в насыщенном состоянии. Т.о., клинические проявления этих нарушений включают аномалии соединительной ткани, аномалии НС и разл. биохим. аномалии, отражающие дисфункцию цикла мочевины. В этом разделе обобщены клинические и лабораторные признаки недостаточной функции Δ1-пирролин-5-карбоксилатсинтазы (см. рис. 9), кодируемой ALDH18A1 (расположенной в 10q24.1), и Δ1-пирролин-5-карбоксилатредуктазы, кодируемой PYCR1 (расположенной в 17q25.3).
Недостаточная активность Δ1-пирролин-5-карбоксилатсинтазы обусловливала несколько фенотипов, в т.ч. синдром де Барси (de Barsy), характеризующийся катарактой, задержкой роста, умственной отсталостью, преждевременным старением (прогероидными признаками) и халазодермией. У некоторых пациентов могут появляться пирамидные признаки. Биопсия кожи позволяет выявить уменьшение размеров эластических волокон и аномалии коллагена. Визуализирующие исследования ГМ указывают на кортикальную атрофию, вентрикуломегалию и снижение уровня креатина.
К лабораторным признакам относятся снижение уровней пролина, орнитина, цитруллина и аргинина, а также умеренная гипераммониемия при анализе натощак. У пациентов могут наблюдаться только периодические отклонения уровней аминокислот в плазме, вероятно, обусловленные временем забора крови по отношению к последнему приему пищи. Интересно, что описаны как АуР, так и АуД-типы наследования. Указанное нарушение можно заподозрить у пациентов с халазодер-мией, задержкой психического развития, легкой гипераммониемией и аминокислотными аномалиями. Диагноз подтверждают методом молекулярного ДНК-анализа/анализа на глутамин в фибробластах кожи при тесте с нагрузкой.
Лечение поддерживающее, хотя для лечения гипераммониемии и церебрального истощения креатина был предложен прием БАД цитруллина/аргинина.
Вредоносные мутации в PYCR1 приводят к аномальной функции митохондриальной Δ1-пирролин-5-карбоксилатредуктазы, которая катализирует последнюю стадию синтеза пролина из Δ1-пирролин-5-карбоновой кислоты. Наиболее постоянные признаки, наблюдаемые у пациентов с доказанными патогенными вариантами PYCR1, включают треугольную форму лица, халазодермию (де Барси-подобный синдром), гиперподвижность суставов, морщинистую кожу, остеодиспластическую геродермию и прогероидные признаки.
При биопсии кожи наблюдается сокращение количества эластических волокон и инфильтрация воспалительными клетками. У некоторых пациентов могут присутствовать эпилепсия, задержка психического и физического развития, умственная отсталость, катаракта и остеопения. Однако во многих семьях с этой патологией имеются близкородственные браки, что усложняет фенотип. Следует отметить, что аминокислотный анализ плазмы обычно не выявляет никаких отклонений от нормы. Диагноз зависит от распознавания кожных признаков и подтверждается молекулярным ДНК-анализом. Имеющиеся родословные семей с PYCR1-ассоциированной патологией подтверждают АуР-тип наследования.