а) Эпидемиология зависимости сердечно-сосудистых заболеваний от уровня липидов крови. Имеется тесная связь между средними показателями потребления насыщенных жиров, ХС плазмы крови и летальностью от ИБС. Ни одно из распространенных хронических заболеваний не имеет такой четкой связи с факторами окружающей среды и генетическими факторами, как ИБС. Это многофакторное нарушение строго коррелирует с увеличением возраста и мужским полом, хотя становится все более явно, что у женщин заболевания сердца остаются недиагностированными. При употреблении табака риск развития указанного заболевания в течение жизни возрастает в 2 раза. Сидячий образ жизни и потребление больших количеств переработанных сахаров, ведущие к ожирению, повышают риск ИБС, увеличивая относительное содержание атерогенных липопротеинов в плазме крови.
Семейный анамнез отражает комбинированное влияние образа жизни и генетической предрасположенности на вероятность развития заболеваний сердца в раннем возрасте.
Риск развития заболеваний сердца в раннем возрасте у пациентов с положительным семейным анамнезом в 1,7 раза выше, чем у пациентов из семей с отрицательным анамнезом.
Атеросклероз начинается в детском возрасте. Результаты исследования прекурсоров Джона Хопкинса (Johns Hopkins Precursors Study) продемонстрировали, что у тех студентов-медиков, у которых уровни ХС крови относились к самому нижнему квартилю, показатели заболеваемости через три десятилетия составили только 10%, а у тех студентов, у которых уровни ХС крови относились к самому высокому квартилю, эти показатели составили 40%. Результаты «Исследования патобиологических детерминант атеросклероза у молодых» продемонстрировали значимую зависимость между абдоминальной жировой массой и распространенностью атеросклероза, по данным аутопсии у лиц в возрасте от 15 до 34 лет. В «Богалузском исследовании сердца» (Bogalusa Heart Study), в котором участвовали >3000 детей негроидной и европеоидной расы, были получены наиболее полные продольные данные, связывающие наличие и тяжесть факторов риска ИБС с полуколичественными показателями выраженности атеросклероза. Атеросклероз коронарных сосудов присутствовал в 8,5% аутопсий, выполненных после смерти в бою или после непреднамеренных травм.
Гипотеза эмбрионального происхождения основана на наблюдении, что у младенцев, рожденных с низкой МТ, отмечаются более высокие показатели ССЗ во взрослом возрасте. Данные эпидемиологических исследований подтверждают идею о том, что условия пренатального и раннего постнатального периода могут влиять на состояние здоровья во взрослом возрасте. Дети с высокой для своего гестационного возраста МТР, матери которых в период беременности страдали СД или ожирением, подвержены повышенному риску развития метаболического синдрома (инсулинорезистентность, СД 2-го типа, ожирение, ИБС). У недоношенных детей грудное вскармливание оказывает долгосрочное (13-16 лет) кардиопротективное воздействие.
У подростков, которые в младенческом возрасте находились на грудном вскармливании, отмечались более низкие показатели концентрации СРБ, а показатели соотношения ЛПНП и ЛПВП были на 14% ниже, чем у тех, которые находились на искусственном вскармливании. Один из механизмов, которые могут определять метаболизм и состав тела взрослого человека, заключается во влиянии питания в раннем возрасте и др. переменных, связанных с образом жизни, на экспрессию генов и эпигенетические факторы.
Второй причиной гиперлипидемии м.б. применение ЛП (циклоспорина, ГКС, изотретиноина, ингибиторов протеаз, алкоголя, тиазидных диуретиков, β-блокаторов, вальпррата) или разл. заболевания (нефротический синдром, гипотиреоз, синдром Кушинга, нервная анорексия, механическая желтуха). Применение психотропных ЛП, в т.ч. антипсихотических ЛС второго поколения, напр. оланзапина, сопровождается дислипидемией, ожирением, а также инсулинорезистентностью.
б) Липиды крови и атерогенез. В большом количестве эпидемиологических исследований продемонстрировано наличие связи между гиперхолестеринемией, выражающейся в виде повышенных показателей общего ХС и ХС ЛПНП крови, с атеросклерозом. С появлением возможности определять субкомпоненты классов липидных частиц, а также маркеры воспаления улучшилось понимание процесса атерогенеза и разрыва бляшек, приводящего к ОКС. Атеросклероз поражает прежде всего КА, однако в патологический процесс могут вовлекаться и аорта, и артерии нижних конечностей, и сонные артерии.
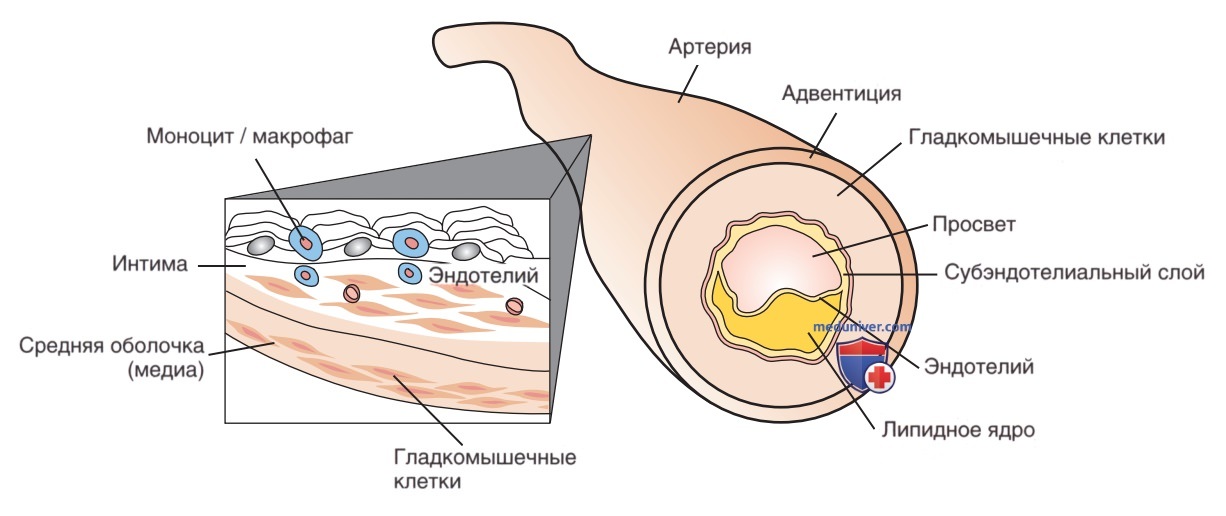
Считается, что атеросклероз начинает развиваться с дисфункции эндотелия сосудов и изменения показателей толщины комплекса интима-медиа. Эти процессы развиваются в предподростковом возрасте при наличии таких факторов риска, как ожирение или семейная гиперхолестеринемия. Сложный процесс проникновения через внутреннюю выстилку сосуда м.б. результатом множества повреждающих факторов, в т.ч. наличия высокотоксичных окисленных частиц ЛПНП. Лимфоциты и моноциты проникают через поврежденный эндотелий, где они становятся макрофагами, накапливают липиды ЛПНП и превращаются в пенистые клетки. Это накопление уравновешивается частицами ЛПВП, которые способны удалять отложения липидов со стенок сосудов. В основе формирования бляшки лежит воспалительный процесс (сопровождающийся повышением СРВ), в котором участвуют макрофаги и стенки артерий. На макроскопическом уровне отложение липидов в субэндотелиальной выстилке артериальной стенки выглядит как полоски жира. Они являются обратимыми в определенных пределах.
Более поздняя стадия развития бляшки включает разрушение гладкомышечных клеток артерий, стимулируемое высвобождаемыми цитокинами и факторами роста. Атерома состоит из ядра, образованного жировой субстанцией и отделенного от просвета сосуда коллагеном и гладкими мышцами (рис. 6).
Рисунок 6. На ранней стадии развитие атеросклероза начинается с проникновения клеток воспаления через внутреннюю выстилку сосуда. Отложение липидов в субэндотелиальной выстилке стенки артерии в конечном счете приводит к разрушению гладкомышечных клеток и формированию атероматозного липидного ядра, которое выпячивается в просвет сосуда. Хронический воспалительный процесс приводит к нестабильности бляшки, создавая тем самым условия для разрыва бляшки и полной окклюзии просвета сосуда формирующимся сгустком
В результате роста атеросклеротической бляшки может возникать ишемия тканей, кровоснабжаемых измененной артерией. Хронический воспалительный процесс в атероме приводит к нестабильности и последующему разрыву бляшки. Адгезия тромбоцитов вызывает формирование сгустка в месте разрыва. В результате, в зависимости от места возникновения тромбоза или тромбоэмболии, развивается ИМ или ОНМК.
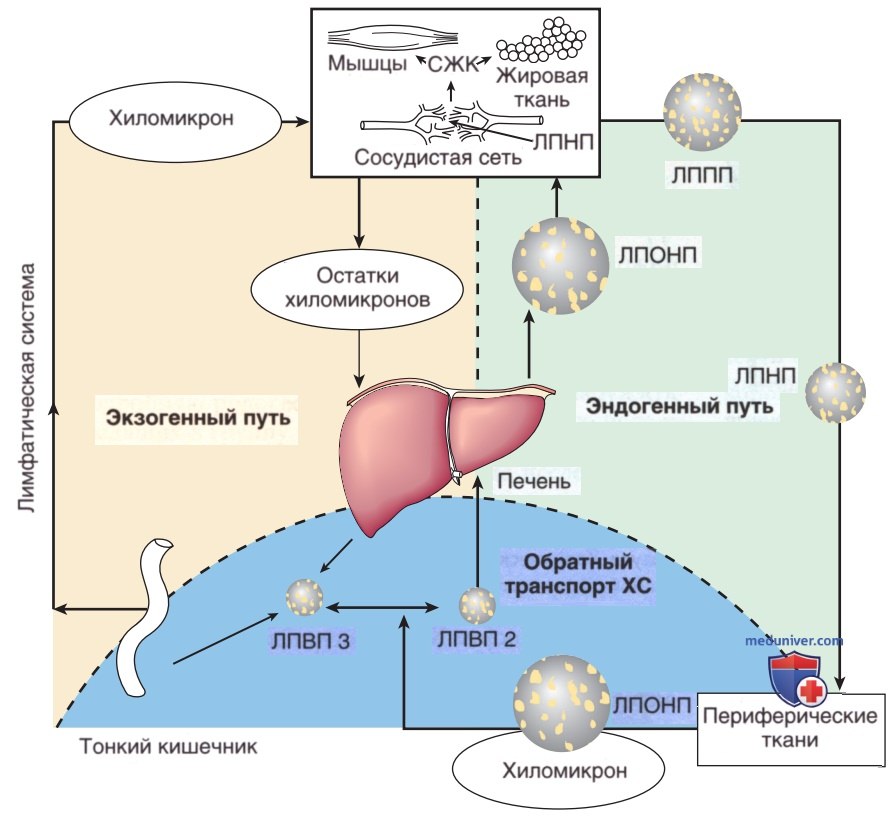
в) Метаболизм и транспорт липопротеинов плазмы. Имеется связь между нарушениями метаболизма липопротеинов и СД и ранним атеросклерозом. Липопротеины — это растворимые комплексы, состоящие из липидов и белков. Они переносят жир, всосавшийся из пищи или синтезированный в печени и жировой ткани, для утилизации и хранения. Жир, содержащийся в пище, транспортируется из тонкого кишечника в виде хиломикронов. Липиды, синтезированные в печени в виде ЛПОНП, катаболизируются до липопротеинов промежуточной плотности (ЛППП) и ЛПНП. ЛПВП играют большую роль в метаболизме ЛПОНП и хиломикронов, а также в транспорте ХС. Неэстерифицированные свободные жирные кислоты представляют собой метаболически активные липиды. Они образуются в результате липолиза триглицеридов, хранящихся в жировой ткани, и циркулируют в плазме крови в связанном с альбуминами виде (рис. 7).
Рисунок 7. Экзогенные, эндогенные и обратные пути метаболизма ХС. Посредством экзогенного пути жир, поступающий с пищей, переносится из тонкого кишечника в периферические ткани и печень в виде хиломикронов. Эндогенный путь заключается в секреции ЛПОНП из печени и их расщеплении до липопротеинов промежуточной плотности (ЛППП) и ЛПНП. Триглицериды, входящие в состав ЛПОНП, под действием липопротеинлипазы (ЛПЛ) подвергаются гидролизу в сосудистой сети. В результате образуются свободные жирные кислоты (СЖК), которые используются или хранятся в мышечной и жировой ткани. Роль метаболизма ЛПВП заключается в переносе избыточных количеств ХС из периферических тканей обратно в печень, где они выводятся с желчью. Вновь образовавшиеся в печени и тонком кишечнике (насцентные) частицы ЛПВП-3 эстерифицируются до более зрелых частиц ЛПВП-2 путем перемещения хиломикронов и ЛПОНП в ядро ЛПВП, происходящего при участии ферментов. Эти частицы удаляются из кровотока путем эндоцитоза
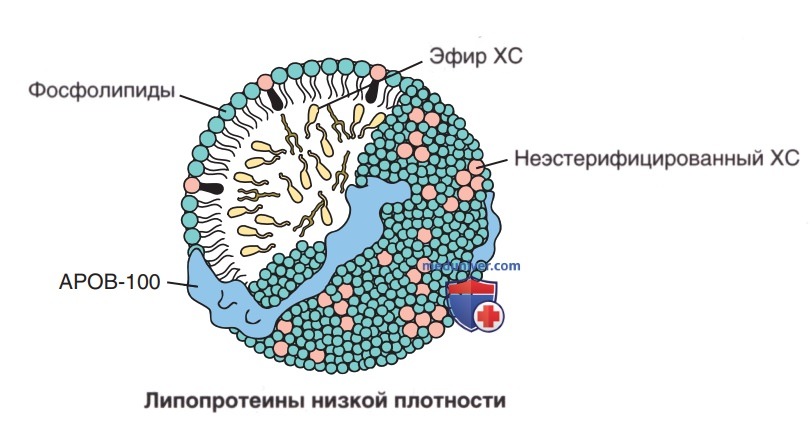
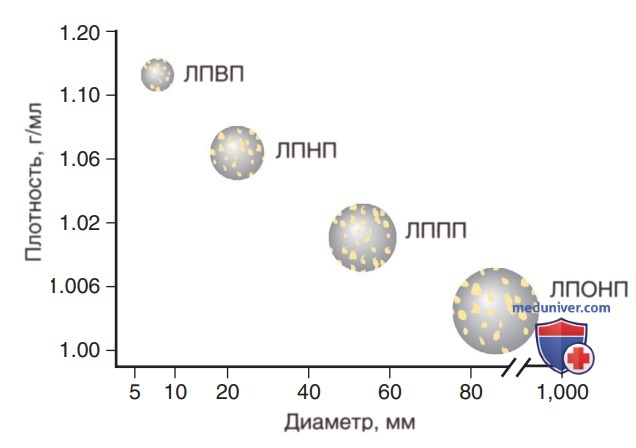
Липопротеины состоят из центрального ядра, которое содержит триглицериды и холестериловые эфиры. Это ядро окружено фосфолипидами, ХС и белками (рис. 8). Плотность липопротеинов, принадлежащих к нескольким классам, обратно пропорциональна соотношению липидов и протеинов. Последние, как правило, плотнее (рис. 9). Липопротеины состоят из центрального ядра, которое содержит триглицериды и эфиры ХС. Это ядро окружено фосфолипидами, ХС и белками.
Рисунок 8. Схематическое изображение липопротеина низкой плотности. Липопротеин состоит из центрального ядра, которое содержит триглицериды и холестериловые эфиры. Это ядро окружено фосфолипидами, ХС и белками
Рисунок 9. Плотность липопротеинов, принадлежащих к нескольким классам, обратно пропорциональна соотношению липидов и белков. Липиды обладают меньшей плотностью, чем белки, поэтому чем больше липидов содержит частица, тем больше ее размер и тем меньше плотность. ЛПВП — липопротеины высокой плотности; ЛПНП — липопротеины низкой плотности; ЛППП — липопротеины промежуточной плотности; ЛПОНП — липопротеины очень низкой плотности
Белки, известные как аполипопротеины, не только выполняют роль структурных белков, но и отвечают за разл. метаболические функции — являются кофакторами или ингибиторами ферментных путей и связываются с рецепторами на поверхности клеток, являясь посредниками липопротеинов (табл. 6). АпоА является основным аполипопротеином (Апо) ЛПВП. АпоВ присутствует в ЛПНП, ЛПОНП, ЛППП и хиломикронах. АпоВ-100 синтезируется в печени, тогда как апоВ-48 — в тонком кишечнике. АпоС-1, С-П и С-Ш — небольшие пептиды, играющие важную роль в метаболизме триглицеридов. Потеря функции и разрушительные мутации гена АРОСЗ сопровождаются низкими уровнями триглицеридов и уменьшают риск ИБС.
Подобным образом АпоЕ, присутствующий в ЛПОНП, ЛПВП, хиломикронах и остатках хиломикронов, играет важную роль в клиренсе триглицеридов.
1. Транспорт экзогенных (поступающих с пищей) липидов. Все поступающие с пищей жиры, за исключением среднецепочечных триглицеридов, эффективно переносятся в кровеносное русло через лимфатические сосуды, дренирующие слизистую оболочку тонкого кишечника. Триглицериды и эфиры ХС соединяются с апоА и апоВ-48 в слизистой оболочке тонкой кишки. В результате формируются хиломикроны, которые через лимфатическую систему переносятся в периферическое сосудистое русло. Частицы ЛПВП доставляют апоС-П к хиломикронам, которые необходимы для активации пипопротеинлипазы (ЛПЛ) в эндотелии капилляров жировой ткани, сердца и скелетных мышц. СЖК окисляются, эстерифицируются, после чего хранятся в виде триглицеридов или высвобождаются в сосудистое русло в связанном с альбуминами виде и переносятся в печень. После гидролиза триглицеридного ядра хиломикрона частицы апоС возвращаются в ЛПВП.
В последующем апоЕ, перенесенный из ЛПВП в остатки хиломикронов, облегчает связывание частицы с рецептором ЛПНП в печени (ЛПНП-Р).
Попадая в гепатоцит, остаток хиломикрона может встраиваться в мембраны, секретироваться обратно в сосудистое русло в качестве липопротеина или секретироваться в виде желчных кислот. В норме все поступившие с пищей жиры распределяются по организму в течение 8 ч после последнего приема пищи. Исключением служат люди с нарушениями метаболизма хиломикронов. Постпрандиальная гиперлипидемия является фактором риска атеросклероза. При нарушении транспорта хиломикронов и их остатков они могут всасываться в стенку кровеносных сосудов в составе пенистых клеток. Такие клетки образуются при поглощении макрофагами эфиров ХС. Это начальная стадия развития жировых полосок.
2. Перенос эндогенных липидов из печени. Эндогенный путь метаболизма липопротеинов включает формирование ЛПОНП в печени и секрецию из нее, а также расщепление до частиц ЛППП и ЛПНП. Жирные кислоты, которые используются в синтезе ЛПОНП в печени, поступают в основном из сосудистого русла. ЛПОНП, по-видимому, выводятся из печени сразу после синтеза и состоят из триглицеридов, эфиров ХС, фосфолипидов и апоВ-100. Вновь образовавшиеся частицы ЛПОНП секретируются в сосудистое русло, объединившись с апоС и апоЕ. Размер частиц ЛПОНП определяется количеством триглицерида. Частицы прогрессивно уменьшаются в размерах по мере того, как триглицерид гидролизуется под действием ЛПЛ. Этот процесс сопровождается образованием СЖК, которые используются или хранятся в мышечной и жировой ткани. При гидролизе 80% триглицерида, имеющегося в частицах ЛПОНП, образуются частицы ЛППП, которые содержат одинаковые количества ХС и триглицерида. Сохранившиеся остатки ЛППП превращаются в ЛПНП и доставляются в периферические ткани или печень.
АпоЕ прикрепляется к остаткам частиц ЛППП, что позволяет им связываться с клетками и в последующем встраиваться в лизосомы.
У лиц с дефицитом апоЕ2 или печеночной триглицеридлипазы ЛППП накапливаются в плазме крови.
У ЗЛ 70% ХС плазмы представлено частицами ЛПНП. Рецепторы к ЛПНП имеются на поверхности почти всех клеток. Основная часть ЛПНП захватывается печенью, а остальное транспортируется в периферические ткани — надпочечники и половые железы — и используется в синтезе стероидов. Дислипидемия в значительной степени зависит от активности ЛПНП-Р. В гомеостазе липидов большую роль играет также эффективность превращения ЛПОНП в ЛПНП. Уровень 50 мг/дл, который является нормой для новорожденных, вероятно, достаточен для синтеза стероидов в течение всего жизненного цикла.
3. Липопротеины высокой плотности и обратный транспорт холестерина. Поскольку секреция липидных частиц из клеток печени в желчь — единственный механизм выведения ХС из организма, перенос избыточных количеств ХС из клеток периферических тканей является жизненно важной функцией ЛПВП. ЛПВП тяжело нагружены липопротеинами, содержащими апоА-I, который, в отличие от липопротеинов В, является неатерогенным. Образующиеся частицы ЛПВП, которые содержат мало ХС и секретируются печенью и тонким кишечником, эстерифицируются до более зрелых частиц ЛПВП-2. Эта реакция проходит с участием фермента лецитинхолестеринацилтрансферазы (ЛХАТ), который облегчает перенос хиломикронов и ЛПОНП в ядро ЛПВП. ЛПВП-2 могут переносить эфиры ХС обратно в апоВ-липопротеины посредством транспортного белка холестериловых эфиров (СЕТР). Кроме того, богатые ХС частицы могут выводиться из плазмы путем эндоцитоза, завершая обратный транспорт ХС.
Низкий уровень ЛПВП может быть связан с генетическими факторами (дефицит апоА-I) или возникать вторично на фоне повышенного уровня триглицеридов плазмы.
Дефицит ЛХАТ приводит к неполному созреванию частиц ЛПВП, что отражается на их способности к обратному транспорту ХС. В результате снижается защитный эффект ЛПВП в отношении атеросклероза. Однако в редких случаях сообщается о меньшей, чем ожидалось, выраженности атеросклероза, несмотря на низкий уровень ЛПВП, возникающий вторично на фоне дефицита ЛХАТ. Данный факт указывает на то, что взаимосвязь, по неизвестным причинам, м.б. разной.
г) Гиперлипопротеинемия:
1. Гиперхолестеринемия. См. табл. 7.
- Семейная гиперхолестеринемия. Семейная гиперхолестеринемия (СГ) — моногенное аутосомно-кодоминантное расстройство, характеризующееся выраженным повышением уровней ЛПНП, ранним развитием ССЗ и развитием ксантом сухожилий. В прошлом семейную гиперхолестеринемию относили к дефектам активности ЛПНП-Р. К этиологическим факторам данного нарушения метаболизма липопротеинов относятся также дефекты генов, кодирующих апоВ [а также пропротеиновой конвертазы субтилизин-кексин типа 9 (PCSK-9; англ. Proprotein convertase subtilisin/kexin type 9)]. Почти все из 1200 описанных мутаций приводят к нарушению синтеза ЛПНП-Р (рецептор-негативные), остальные вызывают нарушение связывания или высвобождения на уровне «липопротеин — рецептор». Рецептор-негативные мутации приводят к развитию более тяжелых фенотипов, чем рецептор-дефективные мутации.
- Гомозиготная семейная гиперхолестеринемия. Гомозиготы с семейной гиперхолестеринемией наследуют два аномальных измененных гена, кодирующих рецепторы к ЛПНП. Это приводит к значительному повышению уровней ХС плазмы (500-1200 мг/дл). Уровни триглицеридов не отклоняются от нормы или незначительно повышены, уровни ЛПВП могут быть незначительно снижены. Это заболевание возникает у 1:500 000 населения. У пациентов с рецептор-негативными мутациями активность ЛПНП-Р составляет <2% от нормы, а у пациентов с рецептор-дефективными мутациями — 25%.
У последних прогноз более благоприятный.
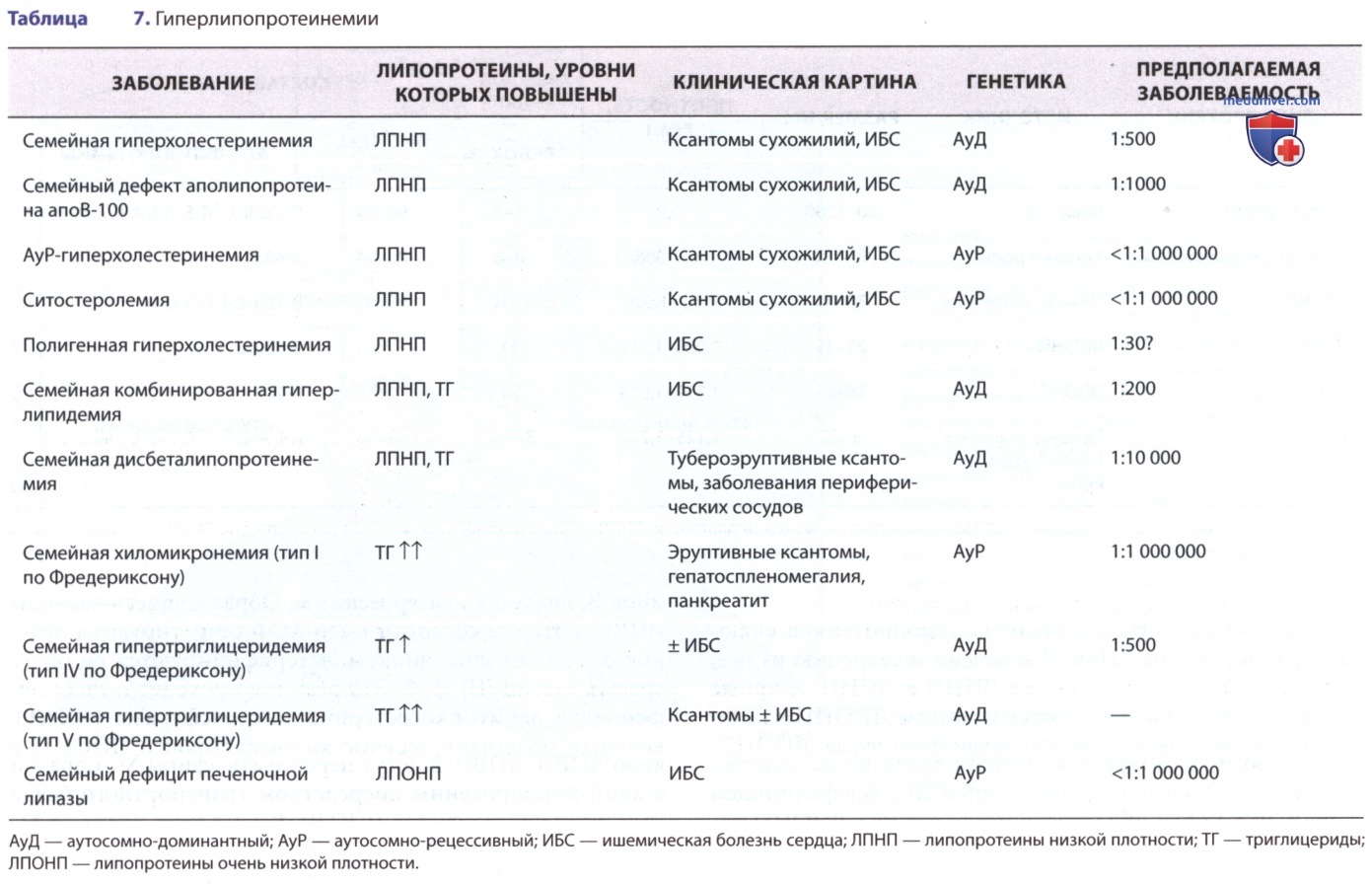
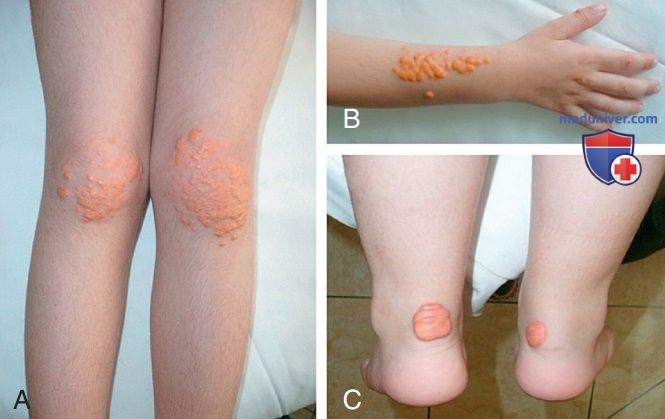
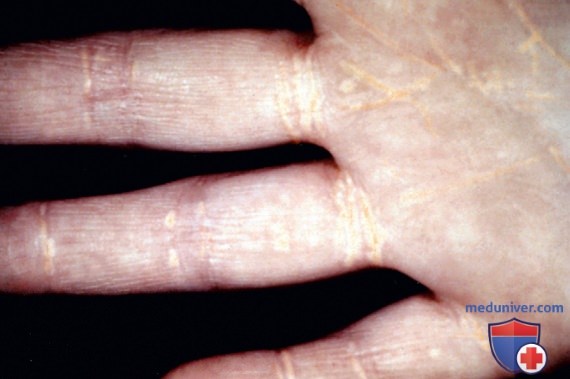
В целом прогноз у пациентов с СГ неблагоприятный, вне зависимости от конкретного вида аберрации ЛПНП-Р. В раннем или среднем периоде детства у таких пациентов имеется тяжелый атеросклероз с вовлечением корня аорты и КА. У таких детей, как правило, имеются ксантомы, которые могут приводить к утолщению ахиллова сухожилия или сухожилий разгибателей кистей. Кроме того, ксантомы могут появляться на коже кистей, в области локтевых и коленных суставов или ягодиц (рис. с 10 по 12). Может присутствовать дуга роговицы. Семейный анамнез не информативен, поскольку высокая частота раннего развития заболеваний сердца отмечается среди родственников обоих родителей. Диагноз можно подтвердить генетическими исследованиями или определением активности ЛПНП-Р в культуре фибробластов кожи. Фенотипическую экспрессию генов при этом заболевании можно также оценить, определяя активность рецептора на поверхности лимфоцитов с помощью методов сортировки клеток.
Рисунок 10. Гомозиготная семейная гиперхолестеринемия. Ксантомы сухожилий у мальчика 5 лет, гомозиготного, с семейной гиперхолестеринемией. Обнаружены в области коленных суставов (а), запястья (б) и ахиллова сухожилия (в)
Рисунок 11. Стриарные ксантомы ладоней
Рисунок 12. Эруптивные ксантомы на разгибательной поверхности предплечья
Без лечения гомозиготные пациенты редко доживают до взрослого возраста. Частые симптомы у таких пациентов — коронарная недостаточность, велика вероятность внезапной смерти. Многим детям рекомендуют проведение афереза ЛПНП с целью выборочного удаления частиц ЛПНП из кровеносного русла, поскольку это замедляет прогрессирование атеросклероза. Трансплантация печени также эффективно снижает уровни ЛПНП-Р, однако нередко возникают осложнения, связанные с иммуносупрессией. Ингибиторы ГМГ-КоА могут быть умеренно эффективны в зависимости от конкретного класса дефекта ЛПНП-Р. Комбинированная терапия с использованием эзетимиба, селективно блокирующего всасывание ХС в кишечнике, как правило, приводит к дальнейшему снижению уровней ЛПНП. Такая терапия в значительной степени заменила терапию секвестрантами желчных кислот.
В ранних клинических исследованиях на фоне применения ингибитора микросомального белка-переносчика триглицеридов ломитапида (ЛП для приема внутрь) отмечали значительное снижение уровней всех апоВ-липопротеинов, в т.ч. ЛПНП, но наличие побочного эффекта в виде отложения жира в печени ограничивает рассмотрение данного фармакологического подхода.
Мипомерсен (ЛП для п/к введения), представляющий собой антисмысловой олигонуклеотид, связывается с последовательностью, которая кодирует аполипопротеин В, снижает синтез апоВ, а следовательно, и синтез ЛПОНП и ЛПНП. Уровни ХС ЛПНП на фоне терапии этим ЛП могут снижаться на 25%. Отмечаются такие побочные эффекты, как гриппоподобные симптомы, стеатоз печени и цирроз печени.
2. Гетерозиготная семейная гиперхолестеринемия. Гетерозиготная семейная гиперхолестеринемия является наиболее распространенной формой единичных генных мутаций, сопровождающихся ОКС и атеросклеротической ИБС у взрослых. Распространенность заболевания составляет 1:250 человек по всему миру, однако в отдельных группах населения, напр. у франко-канадцев, африканцев и у ливанских христиан, этот показатель может быть выше. Это объясняется эффектом основателя — носителя уникальных новых мутаций. Эффект основателя, замеченный у евреев-ашкенази, можно проследить посредством анализа нарушения генетического равновесия до изначальной мутации, возникшей в Литве в 1400-е гг.
Более половины смертей в западных странах связаны с заболеваниями сердца. В патогенезе ИБС имеют значение как факторы окружающей среды, так и генетические факторы. Их сложное взаимодействие определяет фенотипическую экспрессию заболевания.
Поскольку гетерозиготная семейная гиперхолестеринемия является кодоминантным заболеванием с почти полной пенетрантностью, заболевание развивается у 50% родственников первой степени и у 25% родственников второй степени. По оценкам, 20 млн человек по всему миру страдают семейной гиперхолестеринемией. Как правило, средний возраст появления симптомов ИБС составляет 45-48 лет у мужчин и на десять лет позже у женщин. Результаты генетических исследований у людей, полностью отвечающих клиническим критериям диагностики гетерозиготной СГ, не всегда положительные. Это зависит от исследуемой популяции, в т.ч. от того, состоит ли она из детей или из взрослых.
ВОЗ поставила целью разработать индивидуализированные стратегии борьбы, направленные на СГ в связи с огромными показателями заболеваемости и смертности при СГ. Относительно небольшой процент популяции вносит непропорционально большой вклад в бремя ССЗ. Заболевание характеризуется четкой клинической картиной. Существуют эффективные методы лечения.
Невозможно переоценить важность семейного анамнеза при подозрении на СГ, особенно с учетом того, что в учреждениях первичного звена здравоохранения скрининговые исследования уровня ХС проводят у 3% детей. В рекомендациях ААР и Национального института сердца, легких и крови рекомендуется проводить скрининговые исследования уровней ХС всем детям. Однако многие не согласны с этими рекомендациями и не следуют им. Отмечается повышенный интерес к генетическим исследованиям у детей с подозрением на семейную гиперхолестеринемию, поскольку фенотипы варьируют в зависимости от генотипа. Фактически у лиц с семейной гиперхолестеринемией риск ИБС может быть почти в 20 раз выше, чем в общей популяции.
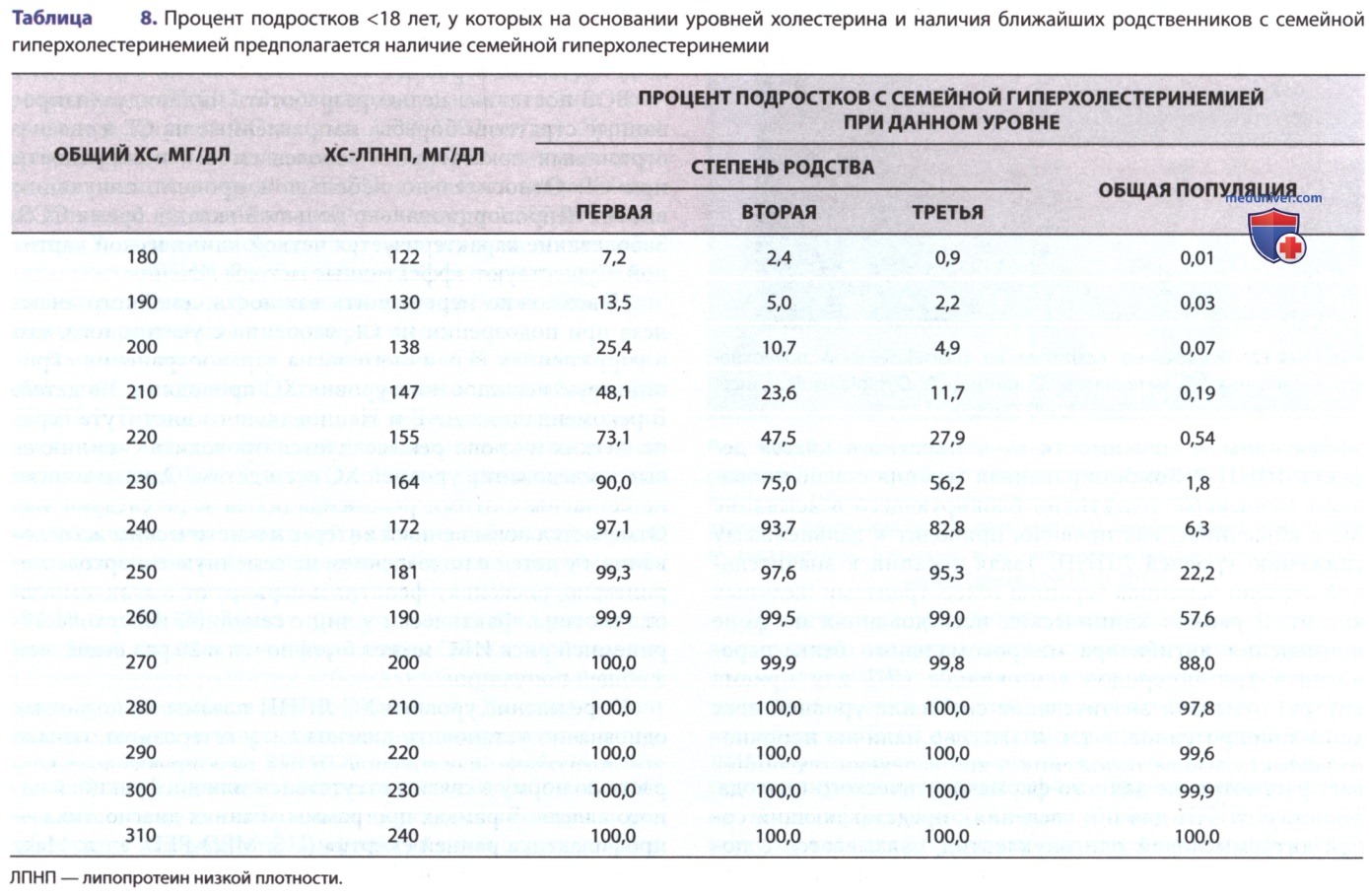
Определение уровней ХС-ЛПНП плазмы не позволяет однозначно установить диагноз СГ у гетерозигот, однако эти показатели, как правило, в два раза превышают возрастную норму в связи с отсутствием или дисфункцией одного аллеля. В рамках программы «Ранняя диагностика — профилактика ранней смерти» (U.S. MED-PED; англ. Make Early Diagnosis-Prevent Early Death) сформулировали ДК. Аналогичные критерии с незначительными отличиями существуют в Великобритании (критерии Саймона Брума) и Голландии (критерии голландской сети липидных клиник). В хорошо известных семьях с СГ диагноз можно с высокой степенью достоверности установить по пороговым значениям ЛПНП. Для установления диагноза у членов ранее не диагностированных семей требуются более строгие критерии: убедительные данные в пользу аутосомного характера наследования и более высокие пороговые значения ЛПНП. При уровне общего ХС 310 мг/дл среди взрослых, принадлежащих к общей популяции, только у 4% будет семейная гиперхолестеринемия, тогда как среди взрослых, являющихся родственниками первой степени человека с установленным диагнозом «семейная гиперхолестеринемия», заболевание будет у 95%.
Показатели математической вероятности семейной гиперхолестеринемии, рассчитанные в рамках программы MED-PED и подтвержденные методами молекулярной диагностики, получены исключительно по данным популяционной когорты США и не применимы к населению др. стран.
При обнаружении высоких уровней ХС у детей следует немедленно провести масштабное обследование взрослых родственников первой и второй степени родства (скрининг на уровень ХС в форме обратного каскада). В общей популяции у ребенка <18 лет при общем уровне ХС плазмы 270 мг/мл и/или ХС-ЛПНП 200 мг/мл вероятность наличия семейной гиперхолестеринемии составляет 88% (табл. 8). Формальный клинический диагноз СГ выставляют при наличии повышенных уровней ХС-ЛПНП (пограничные значения уровней ХС-ЛПНП для 95-го процентиля у детей варьируют в зависимости от возраста и ниже, чем у взрослых; табл. 9) у 2 и более членов семьи. Следовательно, для того чтобы предположить диагноз «семейная гиперхолестеринемия» у ребенка, у родственника которого этот диагноз установлен, достаточно лишь умеренного повышения уровня общего ХС до 220 мг/дл (ХС-ЛПНП — 160 мг/дл; табл. 8). Диагностика семейной гиперхолестеринемии у детей еще больше затрудняется отсутствием клинических стигм, напр. ксантоматоза, которые используются в схемах Саймона Брума и голландской сети липидных клиник, что подчеркивает сдвиг в сторону генетического диагноза.
Лечение детей с семейной гиперхолестеринемией следует начинать со строгой диеты с низким содержанием жиров. Одной диеты редко бывает достаточно для снижения уровней ХС крови до приемлемых значений (ХС-ЛПНП <130 мг/дл). Эзетимиб блокирует всасывание ХС в ЖКТ. Для этого ЛП характерен низкий риск развития побочных эффектов. Данные указывают на то, что эзетимиб снижает уровень общего ХС на 20-30 мг/дл. Ингибиторы ГМГ-КоА-редуктазы (статины) являются ЛП выбора для лечения СГ, поскольку обладают высокой эффективностью и приемлемым профилем риска. Имеется достаточный опыт клинического применения этого класса ЛП у детей >10 лет, позволяющий документально подтвердить, что у детей они так же эффективны, как у взрослых, и что у детей риск повышения уровней ферментов печени и миозита не выше, чем у взрослых. Препараты еще одного класса, ингибиторы PCSK-9, представляют собой моноклональные АТл. Они блокируют действие PCSK-9, что снижает уровень ЛПНП-Р. Эти ЛП повышают уровни ЛПНП-Р; это приводит к значительному снижению уровней ХС-ЛПНП плазмы.
Ингибиторы PCSK-9 эффективны у взрослых, не переносящих статины, и у взрослых с субтерапевтической эффективностью статинов. У детей эти ЛП применяют в качестве экспериментального метода лечения.
- Семейный дефект аполипопротеина апоВ-100. Семейный дефект аполипопротеина апоВ-100 является АуД-заболеванием, не отличимым от гетерозиготной СГ. Уровни ХС ЛПНП повышены, уровни триглицеридов в норме. У взрослых часто развивается ксантоматоз сухожилий и ранняя ИБС. Семейный дефект аполипопротеина апоВ-100 развивается в результате мутации в рецептор-связывающем участке апоВ-100, лиганде рецептора ЛПНП. Предполагаемый показатель у людей, принадлежащих к западной культуре, составляет 1:700 человек. Как правило, причина заключается в замещении глутамина на аргинин в белке апоВ-100 в позиции 3500. В результате снижается способность ЛПНП-Р к связыванию ХС-ЛПНП и нарушается процесс удаления ХС-ЛПНП из сосудистого русла. Специальные методы лабораторных исследований позволяют отличить семейный дефект аполипопротеина апоВ-100 от семейной гиперхолестеринемии, однако за исключением исследовательских целей в этом нет никакой необходимости, поскольку оба заболевания лечат одинаково.
- Аутосомно-рецессивная гиперхолестеринемия. Это редкое заболевание, причина которого заключается в нарушении ЛПНП-Р-опосредованного эндоцитоза в печени, проявляется клинически тяжелой гиперхолестеринемией. При этом показатели ХС занимают промежуточное положение между показателями при гомозиготной и гетерозиготной СГ. Это заболевание особенно распространено среди сардинцев и умеренно отвечает на лечение ингибиторами ГМГ-КоА-редуктазы.
- Ситостеролемия. Редкое АуР-заболевание, характеризующееся избыточным всасыванием в кишечнике растительных стеролов. Ситостеролемия возникает в результате мутаций в системе белка-переносчика, связывающего АТФ (ABCG5 или ABCG8), которая отвечает за ограничение адсорбции растительных стеролов в тонком кишечнике. Небольшое количество адсорбированных растительных стеролов способствует секреции желчи. Уровни ХС плазмы м.б. значительно повышены. Это приводит к образованию ксантом сухожилий и раннему атеросклерозу. Кроме того, отмечаются такие признаки, как гемолитическая анемия, макротромбоцитопения (тромбоциты большого размера, их количество снижено) и кровотечения. Диагноз можно подтвердить повышенными уровнями ситостерола плазмы. Лечение ингибиторами ГМГ-КоА-редуктазы неэффективно, однако отмечается положительный эффект на фоне применения ингибиторов абсорбции ХС, напр. эзетимиба, а также секвестрантов желчных кислот.
- Полигенная гиперхолестеринемия. Первичное повышение уровней ХС-ЛПНП у детей и взрослых чаще всего носит сложное взаимодействие нескольких генетических факторов и факторов окружающей среды (диета), а не дефект какого-то одного гена. Уровни ХС крови умеренно повышены. Уровни триглицеридов не отклоняются от нормы. Полигенная гиперхолестеринемия увеличивается в семьях, ведущих обычный образ жизни, однако развитие этого заболевания не следует прогнозируемому наследственному паттерну, который обнаруживают при моногенных дефектах липопротеинов. Лечение детей с полигенной гиперхолестеринемией направлено на переход к ЗОЖ — сокращение потребления общих и насыщенных жиров, ежедневные физические упражнения продолжительностью не менее 1 ч. Необходимость в применении ЛП, снижающих уровень ХС, возникает редко.
3. Гиперхолестеринемия с гипертриглицеридемией:
- Семейная комбинированная гиперлипидемия. Это АуД-заболевание характеризуется умеренным повышением уровней ХС-ЛПНП, триглицеридов и снижением ХС-ЛПНП плазмы крови. Семейная комбинированная гиперлипидемия (СКГ) является наиболее распространенным первичным нарушением обмена липидов, поражая 1:200 человек. В семейном анамнезе, как правило, имеются случаи ранних заболеваний сердца. Для установления формального диагноза требуется наличие не менее двух родственников первой степени с подтвержденной дислипидемией, относящейся к одному из трех вариантов: 1) уровни ХС-ЛПНП >90-го процентиля; 2) уровни ХС-ЛПНП и триглицеридов >90-го процентиля и 3) уровни триглицеридов >90-го процентиля. Фенотип у одного и того же человека может меняться. Ксантомы для СКГ не характерны. Повышенные уровни апоВ плазмы крови с увеличением небольших плотных частиц ЛПНП подтверждают диагноз.
У детей и взрослых СКГ сопровождается ожирением, АГ и гиперинсулинемией, что указывает на наличие метаболического синдрома.
Для постановки диагноза у взрослых, согласно III Национальной образовательной программе по холестерину (NCEP; англ. National Cholesterol Education Program III), используют шесть основных компонентов: абдоминальное ожирение, атерогенную дислипидемию, АГ, инсулинорезистентность с нарушением толерантности к глюкозе или без него, признаки воспаления сосудов и протромботическое состояние. По оценкам, 30% взрослых, страдающих ожирением, отвечают критериям диагностики метаболического синдрома. Из них 65% составляют люди с СКГ. Особая предрасположенность к этому заболеванию характерна для выходцев с Индостана испанского и южноазиатского происхождения. Официального определения метаболического синдрома у детей не существует. Абсолютные пограничные значения для установления диагноза у детей не определены без учета таких непрерывных переменных, как взросление, половое созревание и расовая/этническая принадлежность.
СКГ и СД-2 типа имеют много общих признаков с метаболическим синдромом. Этот факт указывает на то, что эти нозологические единицы не настолько разные, как считалось изначально. В исследованиях генетических ассоциаций обнаружено большое количество данных в пользу общего генетического происхождения. Возникающее в результате метаболическое перекрытие сопровождается эктопическим накоплением жира и инсулинорезистентностью. Механизмы, связывающие висцеральное ожирение с метаболическим синдромом и СД-2, до конца не ясны. Возможный объединяющий принцип заключается в том, что ожирение приводит к стрессу метаболического ретикулума. В результате подавляется передача сигнала от рецепторов инсулина, в связи с чем развивается инсулинорезистентность и повышается активность воспалительного ответа. Какое отношение вышеуказанные процессы имеют к атерогенезу, неясно.
Предполагают, что гиперхолестеринемия и, с меньшей степенью, гипертриглицеридемия способствуют развитию ССЗ у пациентов с СКГ.
Если включить в логистическую модель признаки метаболического синдрома, то общие этиологические признаки, напр. висцеральное ожирение, становятся явными. Показатели частоты висцерального ожирения с возрастом увеличиваются, а значимость этого признака в качестве фактора риска заболеваний сердца и СД у детей ограничивается относительно небольшим объемом данных. Хотя в исследованиях с участием взрослых определяют окружность талии и наличие интраабдоминального жира на МРТ-изображениях, в педиатрической клинической практике ожирение все еще определяют по ИМТ.
Метаболический синдром наглядно показывает взаимодействие генетических факторов и факторов окружающей среды. Развитие ранних заболеваний сердца у людей с СКГ в значительной степени объясняется генетической предрасположенностью. Неправильный образ жизни, низкое качество питания и недостаточная физическая активность способствуют развитию ожирения и признаков метаболического синдрома.
Одной из важных мер в лечении данной патологии является изменение образа жизни, в т.ч. переход на диету с низким содержанием насыщенных жиров, трансжиров и ХС, а также сокращение потребления рафинированных сахаров. Большое значение имеет повышенное содержание фруктов и овощей в рационе, а также ежедневные физические упражнения в течение 1 ч. Часто бывает нелегко достичь согласия между детьми и их родителями, но этапное продвижение с большей вероятностью приведет к успеху, чем агрессивные стратегии снижения МТ. Очень важно, чтобы в процессе участвовали лица, ухаживающие за ребенком. Уровни триглицеридов плазмы крови, как правило, очень хорошо отвечают на ограничения в диете, особенно на ограничение количества потребляемых сладких напитков. Уровни ХС крови могут снизиться на 10-15%, однако, если показатели ХС-ЛПНП остаются на уровне >160, следует подумать о назначении медикаментозной терапии.
- Семейная дисбеталипопротеинемия (гиперлипопротеинемия типа III). Семейная дисбеталипопротеинемия (СДБЛ) развивается в результате мутаций в гене, кодирующем апоЕ. Под влиянием факторов окружающей среды (напр., высококалорийной диеты с большим содержанием жиров, избыточного потребления алкоголя) возникает смешанный тип гиперлипидемии. У пациентов отмечается тенденция к повышению уровней ХС и триглицеридов плазмы до примерно одинаковой степени. Уровень ХС-ЛПВП, как правило, не отклоняется от нормы, в отличие от др. причин гипертриглицеридемии, сопровождающихся низким уровнем ЛПВП. Этим редким заболеванием страдает — 1:10 000 населения. АпоЕ опосредует удаление остатков хиломикронов и ЛПОНП из кровеносного русла, связывая их с рецепторами на поверхности гепатоцитов. Полиморфный ген АРОЕ экспрессируется в трех изоформах — апоЕ3, апоЕ2 и апоЕ4. Е4 — «нормальный» аллель, присутствующий в большей части популяции.
Изоформа ароЕ2 обладает меньшей аффинностью к рецептору ЛПНП и встречается с частотой 7%. Люди, гомозиготные по апоЕ2/Е2, наиболее распространенной мутации, сопровождающейся развитием СДБЛ, составляют ~1% популяции, однако заболевание проявляется лишь у меньшинства. Для этого требуется наличие предрасполагающих заболеваний, напр. СД, ожирения, заболеваний почек или гипотиреоза. Люди, гомозиготные по апоЕ4/Е4, входят в группу риска по болезни Альцгеймера с поздним началом и деменции, связанной с повторными спортивными травмами головы.
У большинства пациентов с семейной дисбеталипопротеинемией во взрослом возрасте появляются различные ксантомы. Тубероэруптивные ксантомы напоминают небольшие, похожие на гроздья винограда скопления в области коленных суставов, ягодиц и локтевых суставов. Кроме того, в типичных случаях имеется выраженное оранжево-желтое окрашивание ладонных складок (ладонные ксантомы). Атеросклероз часто проявляется заболеваниями периферических сосудов, возникает, как правило, на четвертом или пятом десятке жизни. У детей сыпь м.б. менее выраженной. У них, как правило, имеются предрасполагающие заболевания.
Диагноз семейной дисбеталипопротеинемии устанавливают по результатам электрофореза липопротеинов. При этом наблюдают широкую бета-полоску, содержащую остатки липопротеинов. Непосредственное определение уровней ЛПНП методом ультрацентрифугирования можно выполнить в лабораториях, специализирующихся на исследовании липидов. Диагноз подтверждается при показателях соотношения ЛПОНП/общих триглицеридов >0,30. При наличии явных физических признаков заболевания диагноз можно подтвердить генотипированием АРОЕ на гомозиготность по апоЕ2. Отрицательные результаты не обязательно исключают диагноз СДБЛ, поскольку др. мутации в гене АРОЕ могут приводить даже к более серьезным проявлениям.
Необходимо проводить медикаментозную терапию семейной дисбеталипопротеинемии, чтобы снизить вероятность развития симптоматического атеросклероза во взрослом возрасте. Эффективны ингибиторы ГМК-КоА-редуктазы, никотиновой кислоты и фибратов. Часто семейная дисбеталипопротеинемия хорошо поддается лечению диетой.
4. Гипертриглицеридемии. Семейные нарушения, связанные с липопротеинами с высоким содержанием триглицеридов, включают как распространенные, так и редкие варианты по системе классификации Фредриксона. К таким нарушениям относятся: семейная хиломикронемия (тип I), семейная гипертриглицеридемия (тип IV) и более тяжелая комбинированная гипертриглицеридемия и хиломикронемия (тип V). Кроме того, к подобной комбинированной гиперлипидемии приводит дефицит печеночной липазы.
- Семейная хиломикронемия (гиперлипидемия типа I). Это редкое моногенное нарушение, подобно семейной гиперхолестеринемии, возникает в результате мутаций, воздействующих на клиренс апоВ-содержащих липопротеинов. Дефицит или отсутствие ЛПЛ или ее кофактора апоС-П, который облегчает липолиз под действием ЛПЛ, приводит к значительному повышению уровней хиломикронов плазмы крови с большим содержанием триглицеридов. Уровни ХС-ЛПВП снижены. Эти частицы выводятся с существенной задержкой, так что плазма крови выглядит мутной даже после длительного периода голодания (рис. 13). Хиломикронемия, обусловленная дефицитом ЛПЛ, сопровождается умеренным повышением уровня триглицеридов, чего не наблюдается при дефиците или отсутствии апоС-П. Оба состояния характеризуются АуР-типом наследования, а их частота составляет приблизительно 1 на миллион населения.
Рисунок 13. Напоминающая молоко плазма пациента с болями в животе
Как правило, заболевание проявляется в детском возрасте острым панкреатитом. М.б. эруптивные ксантомы (обильные мелкие папулы) в области верхних конечностей, коленных суставов и ягодиц, а также гепатоспленомегалия.
Диагноз устанавливают на основании результатов исследования липолитической активности триглицеридов. Лечение хиломикронемии заключается в активном сокращении потребления жиров и назначении жирорастворимых витаминов.
Общее потребление жиров можно увеличить за счет среднецепочечных триглицеридов, которые всасываются в портальную венозную систему. Рыбий жир из печени тресковых рыб («Рыбий жир») также может оказывать положительное воздействие.
- Семейная гипертриглицеридемия (гиперлипидемия типа IV). Семейная гипертриглицеридемия (СГТГ) — АуД-нарушение неизвестной этиологии, возникающее с частотой приблизительно 1:500 человек. Заболевание характеризуется повышением уровня триглицеридов плазмы крови >90-го перцентиля (250-1000 мг/дл), часто в сочетании с незначительным повышением уровня ХС плазмы крови и низкими показателями ЛПВП. СГТГ, как правило, не проявляется до взрослого возраста, хотя экспрессируется у 20% пораженных детей. В отличие от СКГ, СГТГ не считается высокоатерогенным заболеванием. Скорее всего, причиной заболевания является нарушение расщепления ЛПОНП или, реже, гиперпродукция этого класса липопротеинов.
Для установления диагноза должен быть по меньшей мере один родственник первой степени с гипертриглицеридемией. СГТГ следует отличать от СКГ и СДБЛ, которые требуют более интенсивного лечения, направленного на профилактику ИБС или заболеваний периферических сосудов. Как правило, ДД можно провести на основании клинических признаков. СГТГ сопровождается более низкими значениями ХС-ЛПНП, однако в сомнительных ситуациях может помочь определение нормальных уровней апоВ при СГТГ.
В некоторых случаях можно обнаружить более тяжелую гипертриглицеридемию, характеризующуюся повышенными уровнями хиломикронов и частиц ЛПОНП (тип V по Фредериксону). Уровни триглицеридов нередко >1000 мг/дл. У детей заболевание наблюдают редко. В отличие от хиломикронемии (тип I по Фредериксону), дефицита ЛПЛ или апоС-П не отмечается. У этих пациентов во взрослом возрасте нередко развиваются эруптивные ксантомы, а у пациентов с гипертриглицеридемией типа IV — нет. Заболевание может проявляться острым панкреатитом. Как и при др. гипертриглицеридемиях, заболевание может обостриться на фоне злоупотребления алкоголем и терапии эстрогенами.
Прежде чем выставлять диагноз СГТГ, необходимо исключить второстепенные причины транзиторной гипертриглицеридемии. К обострению гипертриглицеридемии могут привести: диета с высоким содержанием простых сахаров и углеводов, злоупотребление алкоголем, а также терапия эстрогенами. Подростков и взрослых нужно расспрашивать об избыточном потреблении газированных и др. сладких напитков, поскольку такое состояние часто наблюдают у людей, ежедневно употребляющих сладкие напитки в объеме большой бутылки или нескольких банок по 12 унций (340,194 г). Прекращение такой практики нередко приводит к резкому снижению уровней триглицеридов, а также МТ у людей с ожирением. При стабилизации ИМТ отмечается тенденция к повышению уровней ХС-ЛПВП.
В педиатрической практике гиперлипидемией сопровождаются такие заболевания, как гипотиреоз, нефротический синдром, атрезия ЖВП, гликогенозы, болезнь Ниманна-Пика, болезнь Тея-Сакса, СКВ, гепатит и нервная анорексия (табл. 10). Гиперлипидемия может обостряться на фоне применения определенных ЛП, напр. изотретиноина, тиазидных диуретиков, антипсихотических ЛП второго поколения, гормональных контрацептивов, ГКС, β-блокаторов, иммуносупрессантов, а также ингибиторов протеаз, применяемых для лечения ВИЧ-инфекции.
При лечении триглицеридемии у детей редко возникает необходимость в применении ЛП, даже в тех случаях, когда, несмотря на ограничение потребления жиров, сахаров и углеводов и увеличение физической активности, уровень триглицеридов превышает 1000 мг/дл. Целью терапии у таких пациентов является предотвращение приступов панкреатита. Детям не рекомендуется назначать фибраты (соли фенофиброевой кислоты) и никотиновую кислоту («Ниацин»), широко применяемые у взрослых. Ингибиторы ГМК-КоА-редуктазы обладают разной степенью эффективности в отношении снижения уровней триглицеридов. Относительно данного класса липидснижающих ЛП накоплено значительно больше опыта, подтверждающего их безопасность и эффективность у детей. FDA одобрило применение рецептурных (ловаза, вассепа) и безрецептурных ЛП рыбьего жира в качестве дополнения к диетотерапии при лечении тяжелой гипертриглицеридемии у взрослых.
- Дефицит печеночной липазы. Дефицит печеночной липазы — очень редкое АуР-заболевание, приводящее к повышению и ХС, а также триглицеридов плазмы. Печеночная липаза гидролизует триглицериды и фосфолипиды в остатках ЛПОНП и ЛППП, предотвращая их конверсию в ЛПНП. Отмечается тенденция скорее к повышению, чем к снижению уровней ХС-ЛПВП, что указывает на диагноз. Методом лабораторных исследований, подтверждающим диагноз, является определение активности печеночной липазы в гепаринизированной плазме.
5. Нарушения метаболизма липопротеинов высокой плотности:
- Первичная гипоальфалипопротеинемия. Изолированное снижение уровней холестерина ЛПВП — семейное заболевание, часто характеризуется картиной, предрасполагающей на АуД-путь наследования, однако может возникать независимо от семейного анамнеза. Это заболевание является наиболее распространенным нарушением метаболизма ЛПВП. Оно определяется как уровни ХС-ЛПВП <10-го процентиля для соответствующего пола и возраста при нормальных уровнях триглицеридов и ХС-ЛПНП плазмы. Сопровождается ли это нарушение более быстрым развитием атеросклероза, неизвестно. По-видимому, гипоальфалипопротеинемия связана со снижением синтеза апоА-I и усилением катаболизма ЛПВП. Необходимо исключить второстепенные причины низкого уровня ХС-ЛПВП, напр. метаболический синдром, и редкие заболевания, напр. дефицит ЛХАТ и болезнь Танжера.
- Семейная гиперальфалипопротеинемия. Это необычное заболевание, при котором риск ИБС у членов семьи снижается. Уровни ХС-ЛПВП >80 мг/дл.
- Семейный дефицит аполипопротеина А-I. Мутации в гене апоА-I могут привести к полному отсутствию ЛПВП в плазме крови. Незрелые ЛПВП синтезируются в печени и тонком кишечнике. Свободный ХС из периферических клеток эстерифицируется ЛХАТ. Т.о. формируются зрелые частицы ЛПВП. Для нормального функционирования ЛХАТ в качестве фермента требуется апоА-I. В результате в сосудистом русле накапливается свободный ХС, что в конечном счете приводит к помутнению роговицы, образованию ксантом на подошвах стоп и раннему атеросклерозу. Однако у некоторых пациентов имеются мутации апоА-I, приводящие к очень быстрому катаболизму белка и не сопровождающиеся атерогенезом, несмотря на показатели ХС-ЛПВП 15-30 мг/дл.
- Болезнь Танжера. Показатели ХС-ЛПВП при этом аутосомно-кодоминантном заболевании составляют <5 мг/дл. Причиной являются мутации в гене, кодирующем белок АВСА1. Этот белок облегчает связывание клеточного ХС с апоА-1. Мутации приводят к накоплению свободного ХС в РЭС. Заболевание проявляется гипертрофией небных миндалин, которые имеют отчетливый оранжевый цвет, и гепатоспленомегалией. При накоплении ХС в шванновских клетках может возникать интермиттирующая невропатия. О таком диагнозе следует подумать, если у ребенка увеличенные оранжевые миндалины и значительно повышены уровни ХС-ЛПВП.
- Семейный дефицит лецитинхолестеринацилтрансферазы. Мутации, затрагивающие ЛХАТ, препятствуют эстерификации ХС, тем самым предотвращая формирование зрелых частиц ЛПВП. Это сопровождается быстрым катаболизмом апоА-I. Значительно повышаются уровни свободного циркулирующего ХС плазмы, что приводит к помутнению роговицы и снижению уровней ХС-ЛПВП <10 мг/дл. Частичный дефицит ЛХАТ известен как болезнь «рыбьего глаза». При полном дефиците развивается гемолитическая анемия и прогрессирующая почечная недостаточность в молодом возрасте. Считается, что это редкое заболевание не приводит к раннему атеросклерозу. Лабораторное подтверждение основано на выявлении снижения эстерификации ХС в плазме.
- Дефицит транспортного белка холестериловых эфиров. Мутации, затрагивающие ген СЕТР, локализуются в хромосоме 16y21. Транспортный белок холестериловых эфиров (СЕТР; англ. Cholesteryl ester transfer protein) облегчает перенос липопротеинов из зрелых ЛПВП в ЛПНП и частицы хиломикрона и обратно. Т.о., он является конечным регулятором скорости переноса ХС в печень для выведения с желчью. Ок. половины зрелых частиц ЛПВП-2 удаляются из сосудистого русла напрямую с помощью рецепторов ЛПВП на поверхности клеток печени. Оставшаяся половина холестериловых эфиров ядра ЛПВП обменивается на триглицериды из ядра липопротеинов апоВ (ЛПОНП, ЛППП, ЛПНП) и переносится в печень. Гомозиготный дефицит СЕТР наблюдают в подгруппах японской популяции с экстремально высокими уровнями ХС-ЛПВП (>150 мг/дл).
- Заболевания, сопровождающиеся низкими уровнями холестерина. Нарушения метаболизма апоВ-содержащих липопротеинов и в/клеточного ХС сопровождаются низкими уровнями ХС плазмы.
- Абеталипопротеинемия. Причиной этого редкого АуР-заболевания являются мутации в гене, кодирующем микросомальный белок-переносчик триглицеридов, который необходим для переноса липидов в насцентные хиломикроны в тонком кишечнике и ЛПОНП в печени. Это приводит к отсутствию хиломикронов, ЛПОНП, ЛПНП и апоВ, а также к очень низким уровням ХС и триглицеридов плазмы крови. С раннего возраста отмечается нарушение всасывания жиров, проявляется диареей, задержкой роста, а также мозжечково-спинномозговой дегенерацией, развивающейся на фоне дефицита витамина Е, проявляется исчезновением глубоких сухожильных рефлексов, прогрессирующим до атаксии и спастичности мышц нижних конечностей во взрослом возрасте. Кроме того, у пациентов с абеталипопротеинемией появляется прогрессирующая пигментная ретинопатия, которая сопровождается снижением остроты ночного и цветового зрения в конечном счете приводит к слепоте. На основании неврологических симптомов и ретинопатии можно ошибочно диагностировать атаксию Фридрейха (наследственная атаксия при дефиците витамина Е).
На то, что это не атаксия Фридрейха, указывают нарушение всасывания — синдром мальабсорбции и акантоцитоз в мазке из клеток периферической крови, которые присутствуют при абеталипопротеинемии.
Многие клинические проявления заболевания — результат нарушенного всасывания жирорастворимых витаминов, напр. Е, А и К. Раннее начало терапии витаминами, особенно витамином Е, может значительно замедлить развитие неврологических последствий. В норме витамин Е переносится из тонкого кишечника в печень хиломикронами. Этот процесс зависит от эндогенного ЛПОНП-пути доставки в системный кровоток и периферические ткани. У родителей детей с абеталипопротеинемией уровни липидов и апоВ крови не отклоняются от нормы.
- Семейная гипобеталипопротеинемия. Для семейной гомозиготной гипобетаалипопротеинемии характерны симптомы, очень сходные с симптомами абеталипопротеинемии. Однако путь наследования очень похож на абеталипопротеинемию, но является аутосомнокодоминантным. Причина заболевания — мутации в гене, кодирующем синтез апоВ-100. Это заболевание отличается от абеталипопротеинемии, при которой у родителей пробандов уровни ХС-ЛПВП и апоВ составляют меньше половины нормы. Симптомы или последствия, сопровождающие гетерозиготное заболевание, отсутствуют.
Выборочная неспособность секретировать апоВ-48 в тонком кишечнике приводит к состоянию, напоминающему абеталипопротеинемию или гомозиготную гипобеталипопротеинемию. Нарушение всасывания хиломикронов, иногда называемое болезнью Андерсона, вызывает стеаторею и дефицит жирорастворимых витаминов. Уровни апоВ-100, который в норме секретируется гепатоцитами, при этом заболевании не отклоняются от нормы.
- Синдром Смита-Лемли-Опица. Пациенты с синдромом Смита-Лемли-Опица (SLOS; англ. Smith-Lemli-Opitz syndrome) часто имеют множественные врожденные аномалии и задержку развития по причине низкого уровня ХС в плазме и накопленных предшественников (табл. 11, 12). При анализе семейной родословной обнаружено, что для этого заболевания характерен АуР-тип наследования. В результате мутаций в гене DHCR7 развивается дефицит микросомального фермента 7-дегидрохолестерин-Δ7-редуктазы (DHCR7; англ. 7-Dehydrocholesterol reductase), который необходим для завершения заключительного этапа синтеза ХС. Каким образом нарушение синтеза ХС приводит к врожденным мальформациям, неизвестно, однако, поскольку ХС является основным компонентом миелина и участвует в передаче сигнала в процессе развития НС, дефекты его синтеза приводят к тяжелым нарушениям развития НС. Предполагаемый показатель заболеваемости SLOS среди людей европеоидной расы составляет 1:20 000-60 000 родившихся.
Среди представителей испаноязычного населения этот показатель несколько выше, а среди людей африканского происхождения — ниже.
Отмечаются случаи самопроизвольных выкидышей плодов с SLOS. SLOS типа II нередко приводит к летальным исходам в конце неонатального периода. При уровнях холестерина плазмы <20 мг/дл выживание невозможно. При проведении лабораторных исследований следует использовать метод газовой хроматографии, поскольку стандартные методы определения уровней липопротеинов включают определение предшественников ХС, что может привести к л/п-результату. Более легкие случаи могут не проявляться до старшего детского возраста. Фенотипические проявления варьируют от микроцефалии, мальформаций сердца и ГМ, полиорганной системной недостаточности до легких признаков дисморфизма и незначительной задержки развития. Лечение включает применение БАД, содержащих ХС (яичный желток), и ингибиторов ГМГ-КоА-редуктазы с целью предотвращения синтеза токсических предшественников проксимальнее ферментного блока.
6. Нарушения внутриклеточного метаболизма холестерина:
- Церебротендинозный ксантоматоз (сухожильномозговой ксантоматоз). Данное АуР-нарушение проявляется клинически в старшем подростковом возрасте ксантомами сухожилий, катарактами и прогрессирующей нейродегенерацией. Причиной заболевания является накопление промежуточных метаболитов желчных кислот со сдвигом в сторону холестанола в результате мутаций в гене, кодирующем стерол-27-гидроксилазу. Этот фермент необходим для нормального митохондриального синтеза желчных кислот в печени. Раннее начало терапии ЛП хенодезоксихолевой кислоты снижает уровни ХС и предотвращает развитие симптомов.
- Болезнь Вольмана и болезнь накопления эфиров холестерина. Это АуР-заболевание развивается в результате дефицита лизосомной кислой липазы. После попадания в клетку в результате эндоцитоза холестерин ЛПНП доставляется в лизосомы, где гидролизуется лизосомной липазой. Нарушение гидролиза при полном отсутствии фермента приводит к накоплению эфиров ХС в клетках. Клинические проявления в виде гепатоспленомегалии, стеатореи и задержки физического развития появляются в раннем младенческом возрасте, смерть наступает в возрасте 1 года. Болезнь накопления эфиров ХС, менее тяжелая форма, чем болезнь Вольмана, показатели активности кислой липазы низкие, но все же определяются.
- Болезнь Ниманна-Пика, тип С. Это нарушение в/клеточного транспорта ХС характеризуется накоплением холестерина и сфингомиелина в ЦНС и ретикулоэндотелиальной системе.
Это мультисистемное заболевание, клинические проявления включают в себя висцеральные (холестаз, спленомегалию или гепатоспленомегалию, инфильтраты в легких, асцит и др.), неврологические (мозжечковую атаксию, дистонию, дисфагию, окуломоторные нарушения, катаплексию, эпилептические приступы, нейросенсорную тугоухость, периферическая невропатия, когнитивные нарушения) и психиатрические симптомы (психоз, кататонию, нарушения обучения, расстройство экспрессии речи, синдром дефицита внимания).
P.S. * КР РФ 2019 г. «Болезнь Ниманна-Пика, тип С».
Летальный исход при этом заболевании, как правило, возникает в подростковом возрасте.
В 10% случаев при молниеносной форме смерть может наступать на первом году жизни.
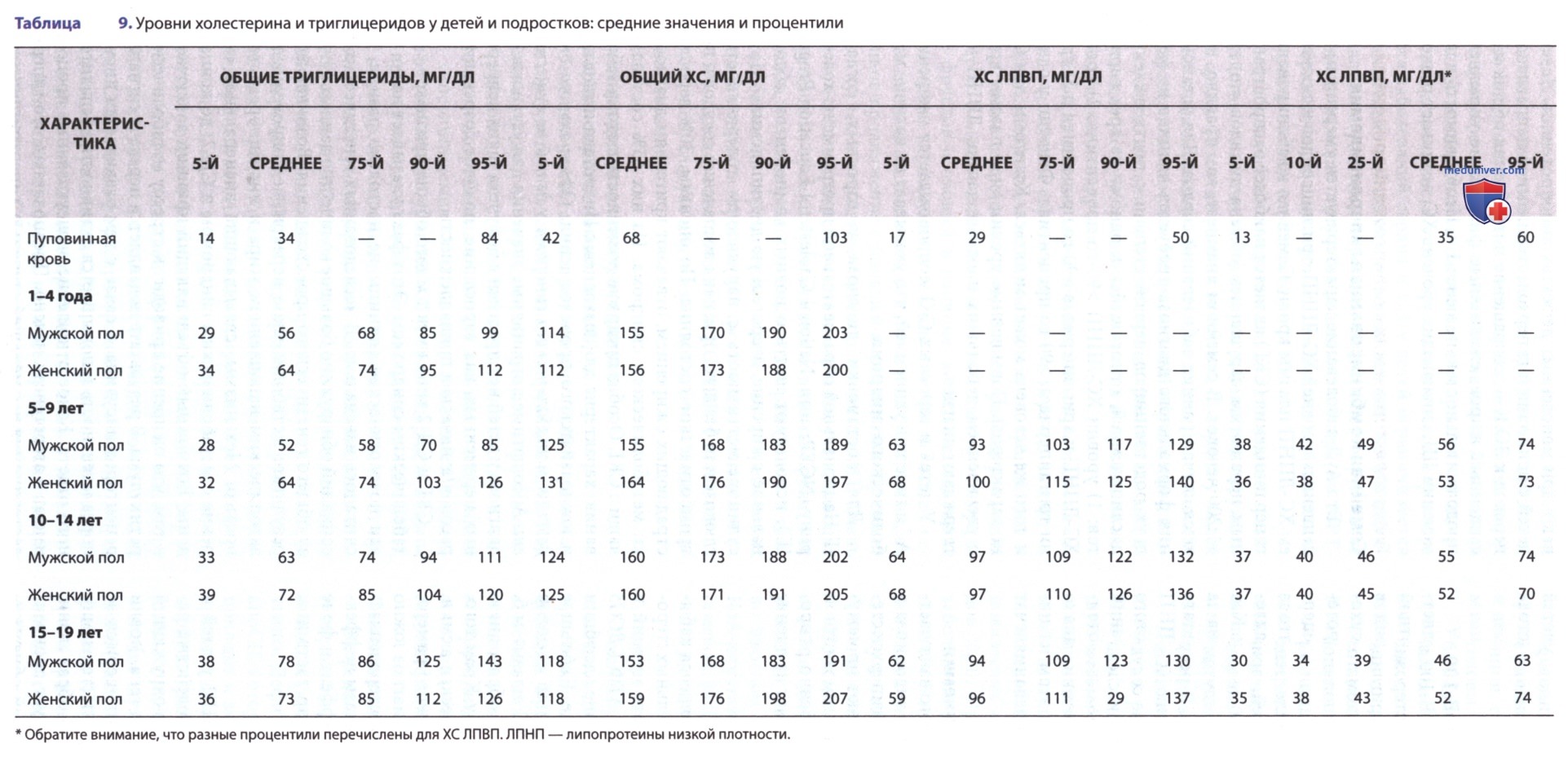
7. Уровень липопротеинов у детей и подростков. В табл. 9, основанной прежде всего на материалах Популяционных исследований клиник по исследованию липидного обмена (Lipid Research Clinics Population Studies), показано распределение уровней липопротеинов у молодых американцев разных возрастов. Уровни общего ХС плазмы быстро нарастают со среднего показателя 68 мг/дл после рождения примерно в два раза к концу неонатального периода. Уровень общего ХС постепенно нарастает до начала полового созревания, когда средние показатели достигают 160 мг/дл. В период пубертата уровни общего ХС временно снижаются. У мальчиков это связано с небольшим снижением уровней ХС-ЛПВП, а у девочек — с незначительным снижением уровней ХС-ЛПНП. Достаточно хорошо прослеживается изменение показателей ХС в зависимости от возраста.
Отмечается тенденция к высоким показателям ХС у членов одной семьи. Это отражает влияние генетических факторов и факторов окружающей среды.
Для детей и подростков приемлемые показатели общего ХС составляют <170 мг/дл, пограничные — 170-199 мг/дл, высокие — >200 мг/дл. Приемлемый уровень ХС-ЛПНП — <110 мг/дл, пограничный — 110-129 мг/дл и высокий — >130 мг/дл. Уровень ХС-ЛПВП должен быть >40 мг/дл.
8. Скрининг уровня холестерина крови. С 2011 г. ААР рекомендует проводить скрининг уровня ХС крови всем детям с использованием единого подхода. Липидный профиль следует определять у всех детей в возрасте с 9 до 11 лет, а затем еще раз в возрасте с 17 лет до 21 года, поскольку после окончания пубертатного периода показатели ХС могут изменяться. Однако если ребенок соответствует выборочным критериям предшествующих рекомендаций, основанных на оценке рисков (ранняя ИБС у родителей или дедушек и бабушек, уровни ХС у родителей >240 мг/дл), скрининг можно провести и в 2 года. Кроме того, данные указывают на то, что определение липидного профиля не натощак м.б. не менее эффективно в плане выявления тяжелых наследственных дислипидемий, как и натощак, так что его можно применять в качестве скринингового исследования первой линии у детей.
Кроме того, определение липидного профиля натощак можно проводить в зависимости от предпочтений родителей, ребенка и врача, особенно в случае опасений по поводу гипертриглицеридемии, поскольку голодание больше отражается на уровне триглицеридов. При отклонении показателей липидной панели от нормы исследования необходимо повторить, особенно при отклонении показателей триглицеридов. Повторное исследование проводят не раньше чем через 2 нед натощак. Для назначения терапии, кроме изменения образа жизни, однократного определения липидного профиля недостаточно.
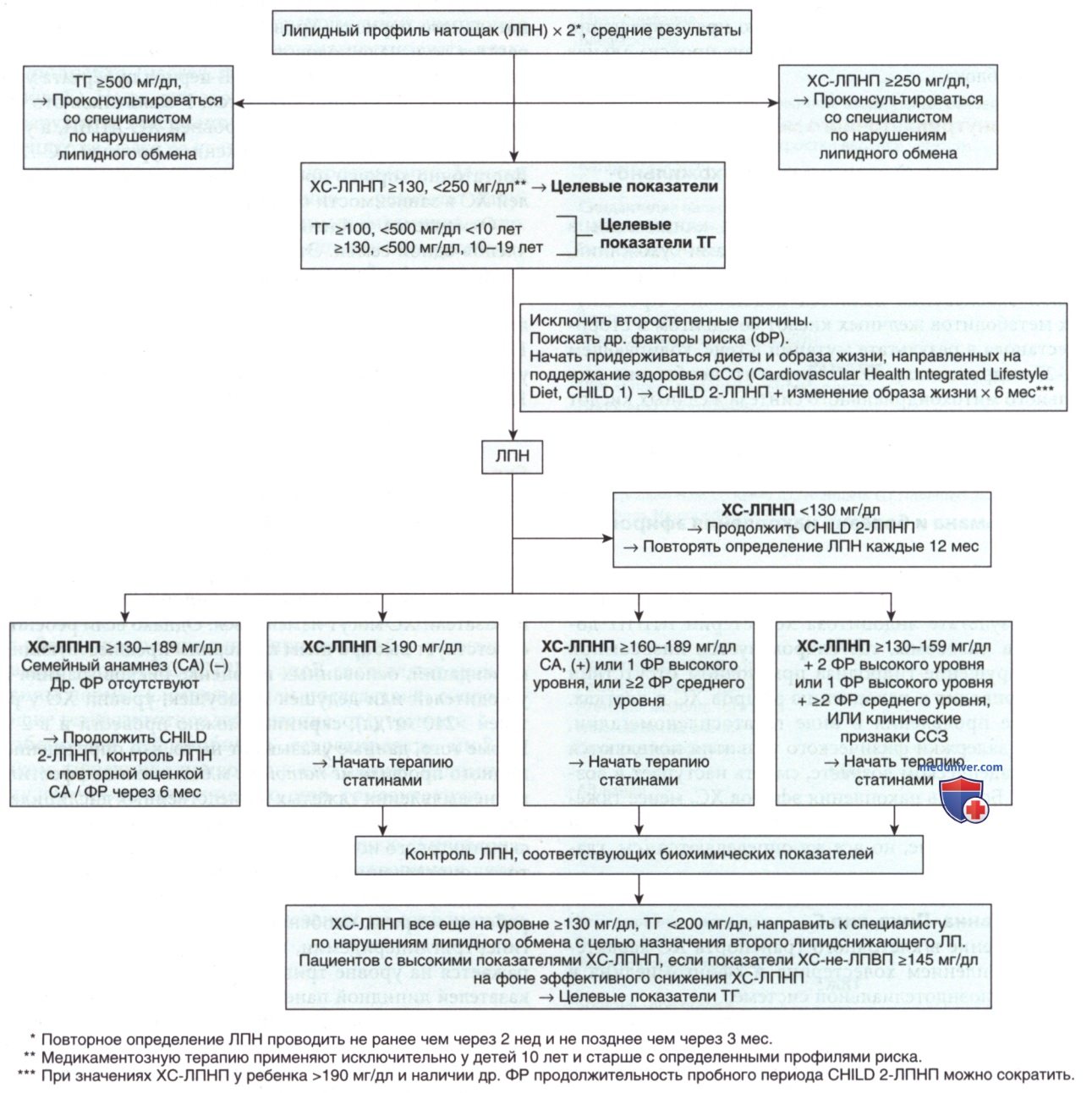
- Оценка рисков и лечение гиперлипидемии. NCEP рекомендует применять ко всем детям популяционный подход, основанный на ведении ЗОЖ, и индивидуальный подход — к детям из группы высокого риска (рис. 14). ААР рекомендует уделять основное внимание поддержанию ЗОЖ, а не агрессивным подходам к снижению МТ.
Рисунок 14. Алгоритм лечения дислипидемии: целевые показатели ХС-ЛПНП (холестерин липопротеинов низкой плотности). Примечание: показатели представлены в мг/дл. Для перевода в систему СИ результаты для общего холестерина (ОХ), холестерина липопротеинов низкой плотности (ХС-ЛПНП), холестерина липопротеинов низкой плотности (ХС-ЛПНП), холестерина липопротеинов высокой плотности (ХС-ЛПВП) и ХС-не-ЛПВП разделите на 38,6; результаты для триглицеридов (ТГ) разделите на 88,6.
Для того чтобы определить оптимальную схему лечения, детей с дислипидемиями делят на группы в зависимости от наличия факторов риска высокого или среднего уровня.
К факторам риска высокого уровня относятся АГ, требующая медикаментозной терапии (показатели АД >99-го процентиля + 5 мм рт.ст.), курение сигарет на данный момент, ИМТ на уровне >97-го процентиля, наличие СД-1/СД-2, хронические заболевания почек, состояние после ортотопической трансплантации сердца и/или болезнь Кавасаки с наличием аневризмы.
К факторам риска среднего уровня относятся АГ, не требующая медикаментозной терапии, ИМТ >95-го процентиля, но <97-го процентиля, уровень ХС-ЛПВП <40 мг/дл, болезнь Кавасаки с регрессировавшими аневризмами КА, хронические воспалительные заболевания, ВИЧ-инфекция и/или нефротический синдром.
У детей лечение дислипидемии всегда начинают с пробного изменения образа жизни, т.е. улучшения характера питания и увеличения физической активности. Это продолжается в течение 6 мес. Избыточная МТ особенно повышает риск ССЗ из-за наличия строгой корреляции с инсулинорезистентностью (метаболический синдром). Хотя стандартизованного определения метаболического синдрома для подростков не существует, вероятно, у половины всех детей с ожирением тяжелой степени имеется инсулинорезистентность. Согласно данным проекта CARDIAC, у 49% пятиклассников при наличии ожирения в сочетании с гиперпигментированной сыпью, акантокератодермией, в соответствии с определением, которое традиционно применяется для взрослых, имеется три и более фактора синдрома инсулинорези-стентности, в т.ч. подтвержденная инсулинорезистентность, АГ, уровень ХС-ЛПВП <40 мг/дл и уровень триглицеридов >150 мг/дл.
Первый этап диеты и образа жизни, направленных на поддержание здоровья ССС (CHILD-1), — первый уровень изменений в диете. Его следует рекомендовать всем детям с дислипидемиями. Диета CHILD-1 разработана специально для детей с факторами риска ИБС. В ней основное внимание уделяется ограничению потребления ХС до 300 мг/сут, ограничению потребления сахаросодержащих напитков, употреблению обезжиренного молока или молока с пониженным содержанием жиров, отказу от пищи с высоким содержанием трансжиров, ограничению продуктов с высоким содержанием натрия и поощрению потребления продуктов с высоким содержанием пищевых волокон. Конкретные рекомендации зависят от возраста ребенка.
Второй этап диеты и образа жизни, направленных на поддержание здоровья ССС (CHILD-2), рекомендуется применять в случаях, когда CHILD-1 оказалась неэффективной. Диета CHILD-2 по многим аспектам напоминает CHILD-1, однако направлена на конкретные типы дислипидемии. Диету CHILD-2 ЛПНП рекомендуют детям с повышенными уровнями ЛПНП, а диету CHILD-2 ТГ — детям, у которых повышены уровни триглицеридов. Основные рекомендации по потреблению калорий согласно диете CHILD-2 заключаются в следующем: на жиры приходится только 25-30% калорий, на насыщенные жиры — <7% калорий, на мононенасыщенные жиры — 10% калорий, потребление ХС составляет <200 мг/сут. Если рекомендуют диету CHILD-2 ЛПНП, акцентируют внимание на применении растительных стеролов и водорастворимых пищевых волокон. Если рекомендуют диету CHILD-2 ТГ, особое внимание уделяют употреблению больших количеств омега-3-жирных кислот и сложных, а не простых углеводов.
Соблюдение указанных рекомендаций по диете обеспечивает количество калорий, достаточное для оптимального роста и развития и не способствующее ожирению. Соблюдение рекомендаций представляет сложную задачу для детей и лиц, ухаживающих за ними. Дети перенимают пищевые привычки от своих родителей. Вероятность успешного перехода на более ЗОЖ значительно повышается, если приемы пищи и перекусы дома организуются с учетом потребностей всей семьи, а не только ребенка. Желательно принимать пищу всей семьей в одно и то же время. В некоторых случаях бабушкам и дедушкам, а также др. лицам, ухаживающим за ребенком и не являющимся его родителями, следует напоминать о том, что не нужно баловать ребенка, который соблюдает диету. Кроме того, возрастание количества детей с ожирением вынуждает некоторые школьные округа ограничивать доступность сладких напитков и предлагать в столовых более полезные блюда.
Кроме того, важной частью начальных изменений образа жизни является изменение привычек в отношении физической активности. Национальная ассоциация спорта и физкультуры рекомендует детям выделять на физические упражнения, соответствующие возрасту, >60 мин/сут большую часть дней недели. Не рекомендуются длительные (>2 ч) периоды отсутствия физической активности, напр. проведение перед телевизором и экранами различных устройств >2 ч.
9. Медикаментозная терапия. См. табл. 13, 14.
У детей, которые не отвечают на строгое соблюдение рекомендаций по изменению образа жизни в течение 6 мес, основным методом лечения является медикаментозная терапия холестеринснижающими препаратами. Вариант назначения лекарственной терапии следует рассмотреть в случае соблюдения следующих условий (см. также на рис. 14).
• Показатели ХС ЛПНП сохраняются на уровне >190 мг/дл.
• Показатели ХС ЛПНП сохраняются на уровне >160 мг/дл при наличии одного фактора риска высокого уровня и/или по меньшей мере двух факторов риска среднего уровня.
• Показатели ХС ЛПНП остаются на уровне >130 мг/дл при наличии по меньшей мере двух факторов риска высокого уровня, одного фактора риска высокого уровня и по меньшей мере двух факторов риска среднего уровня или подтвержденной ИБС.
Ингибиторы ГМГ-КоА-редуктазы, известные также как статины, обладают высокой эффективностью, снижая уровни ХС ЛПНП и уменьшая выраженность воспалительного процесса в бляшках. Тем самым они уменьшают вероятность внезапной коронарной смерти у взрослых из группы риска через несколько недель от начала применения. Механизм действия данного класса препаратов заключается в блокировании биосинтеза ХС в печени. Т.о. стимулируется продукция большего количества рецепторов к ЛПНП на поверхности клеток и облегчается захват ХС-ЛПНП из сосудистого русла.
В панели лечения взрослых NCEP рекомендуется агрессивно снижать уровень ЛПНП до значений <70 мг/дл у пациентов с установленным диагнозом ИБС. На эту информацию следует обратить внимание, поскольку ребенок, отвечающий критериям, по которым принимают решение о назначении холестеринснижающих ЛП, почти всегда наследует свое заболевание от одного из родителей. Нередко при лечении детей возникает вопрос о скрининговом обследовании и лечении родителей или бабушек и дедушек. У детей статины обладают такой же эффективностью. При необходимости они могут снижать уровни ХС-ЛПНП на 50%. Эти ЛП рассматривают в качестве терапии первой линии у детей, соответствующих критериям назначения ЛП. Кроме того, на фоне применения статинов отмечается умеренное снижение уровня триглицеридов и несоответствующее повышение уровня ХС-ЛПВП.
ЛП следует назначать с учетом его профиля побочных эффектов, которые представлены в основном нарушением функции печени и, в редких случаях, рабдомиолизом со вторичной почечной недостаточностью. Не существует данных, свидетельствующих о более частом развитии осложнений у детей, чем у взрослых. Дискомфорт со стороны скелетных мышц у детей встречается реже. Кроме того, может возникнуть лекарственное взаимодействие с др. ЛП, поэтому, чтобы избежать побочных эффектов, необходимо уделить особое внимание ЛП, которые ребенок получает на данный момент. У детей важно регулярно отслеживать уровни печеночных ферментов. При появлении болей в мышцах или слабости следует определить показатели КФК. Можно продолжать применение ЛП до тех пор, пока показатели печеночных (мышечных) ферментов не превысят норму более чем в три раза.
Предполагается, что существует связь между применением статинов и повышенным риском развития СД-2 типа у взрослых, однако у детей эти результаты не оценивались. У детей, получающих статины, определяли уровни половых гормонов, и оказалось, что они не изменяются. Следует еще раз подчеркнуть, что дети, у которых уровни ХС повышены умеренно, как это наблюдается при полигенной гиперхолестеринемии, как правило, не являются кандидатами на применение статинов. Это объясняется профилем побочных эффектов данной группы препаратов и ответом детей на изменение образа жизни. Терапию статинами следует начинать с самой низкой эффективной дозы. Максимальный эффект достигается не ранее чем через 8 нед. Если не удалось достичь целевых показателей ЛПНП, которые у детей, получающих лечение, составляют, как правило, <130 мг, то можно титровать дозу в сторону повышения, тщательно контролируя побочные эффекты.
Др. холестеринснижающие ЛП, напр. никотиновую кислоту и фибраты, у детей применяют намного реже, чем секвестранты желчных кислот и статины. Никотиновую кислоту и фибраты выборочно применяют у детей с выраженной гипертриглицеридемией (>500 мг/дл), которые входят в группу риска по острому панкреатиту, хотя ограничение потребления сложных сахаров (с упором на исключение сладких сахаросодержащих напитков) и углеводов, как правило, приводит к значительному снижению уровней триглицеридов. В действующих рекомендациях предлагают в первую очередь и после достижения целевых показателей ЛПНП сосредоточиться на снижении уровней ХС-ЛПНП. Затем, если показатели триглицеридов будут оставаться на уровне 200-499 мг/дл, а ХС не-ЛПВП — на уровне >145 мг/дл, показана медикаментозная терапия, направленная на снижение уровней триглицеридов.
Пищевые добавки, содержащие омега-3 жирные кислоты, которые выпускаются как в форме безрецептурных ЛП, так и в формах, отпускаемых по рецепту, безопасны и эффективны. Считается, что они снижают уровни триглицеридов, уменьшая их синтез в печени. Недавно считалось, что показатели ХС-ЛПНП у взрослых ок. 70 мг/дл сопровождаются уменьшением атероматозных бляшек в КА и обратным развитием ИБС. Знания в этой области продолжают эволюционировать.
Доказана польза применения эзетимиба у детей, поскольку этот препарат эффективен и обладает благоприятным профилем побочных эффектов. Эзетимиб снижает уровни ХС-ЛПНП плазмы, блокируя всасывание стерола энтероцитами. Этот ЛП продается в качестве дополнения к статинам. Его используют у взрослых, когда на фоне применения только статинов не удается в достаточной степени снизить уровни липидов крови. Достаточное количество сообщений, документально подтверждающих его эффективность при отсутствии побочных эффектов, свидетельствует в пользу того, чтобы при обнаружении умеренной гиперхолестеринемии или при наличии у родителей опасений, затрудняющих применение статинов, рекомендовать применение эзетимиба вместо статинов.
В РФ медикаментозную гиполипидемическую терапию у детей рекомендовано начинать с 8-10-летнего возраста, также на фоне соблюдения диеты. Рекомендуемый целевой уровень ХС ЛНП у детей 8-10 лет — <4,0 ммоль/л, у детей >10 лет — <3,5 ммоль/л. Начинать медикаментозную терапию следует с назначения статинов в низких дозах, постепенно титруя их до оптимальных доз. Если ХС ЛНП без лечения >13 ммоль/л, терапия начинается с назначения статинов в максимально переносимой дозе, при отсутствии желаемого эффекта к статинам добавляют эзетимиб и/или ингибиторы PCSK9 (эволокумаб у детей >12 лет п/к 140 мг каждые 2 нед или 420 мг один раз в месяц). В особо тяжелых случаях к медикаментозной терапии рекомендуется добавить экстракорпоральные методы лечения (иммуносорбцию ЛНП или каскадную плазмофильтрацию)*.
P.S. * Диагностика и коррекция нарушений липидного обмена с целью профилактики и лечения атеросклероза. Российские рекомендации, VII пересмотр.