GM2-ганглиозидоз у ребенка - кратко с точки зрения педиатрии
К GM2-ганглиозидозам относятся болезнь Тея-Сакса и болезнь Сандхоффа; оба заболевания обусловлены снижением активности β-гексозаминидазы и накоплением GM2-ганглиозидов в лизосомах, особенно в ЦНС. В зависимости от возраста начала болезни и клинических проявлений при развитии GM2-ганглиозидозов выделяют инфантильную, ювенильную и взрослую формы. Существует две изоформы β-гексозаминидазы: β-гексозаминидаза А, состоящая из 1-α и 1-β-субъединиц, и β-гексозаминидаза В, состоящая из 2-β-субъединиц.
Дефицит β-гексозаминидазы А возникает в результате мутаций в субъединице а и вызывает болезнь Тея-Сакса, а мутации в субъединице β приводят к дефициту и β-гексозаминидазы А и В и вызывают болезнь Сандхоффа. Оба заболевания наследуются по АуР-типу. Болезнь Тея-Сакса чаще встречается в популяции евреев-ашкенази (носители определяются с частотой 1:25). Частота встречаемости GM2-ганглиозидоза, тип I составляет от 1:3000 новорожденных среди евреев-ашкенази и 1:320 000 новорожденных в США.
Идентифицировано >50 мутаций. Большинство из них связано с инфантильной формой заболевания. У носителей болезни Тея-Сакса, принадлежащих к популяции евреев-ашкенази, три мутации составляют >98% мутантных аллелей, включая одну аллель, связанную с взрослой формой. Мутации, вызывающие подострую или взрослую формы, приводят к образованию ферментных белков с остаточной ферментативной активностью, уровень которой коррелирует с тяжестью заболевания. Также выделяют GM2-ганглиозидоз, вариант В1, который ассоциирован с образованием неактивного изофермента гексозаминидазы А с нарушением гидролиза GM2-ганглиозида.
Известен ряд мутаций, представленный в международной базе OMIM, которые приводят к развитию такого варианта заболевания. Клинические проявления GM2-ганглиозидоза, вариант В1 сходны с клинической картиной ювенильной формы болезни Тея-Сакса*.
P.S. * Журкова Н.В., Вашакмадзе Н.Д., Суханова Н.В. и др. GM2-ганглиозидоз, тип I (болезнь Тея-Сакса) в практике педиатра // Педиатрическая фармакология. 2020. Т. 17. № 6. С. 529-535
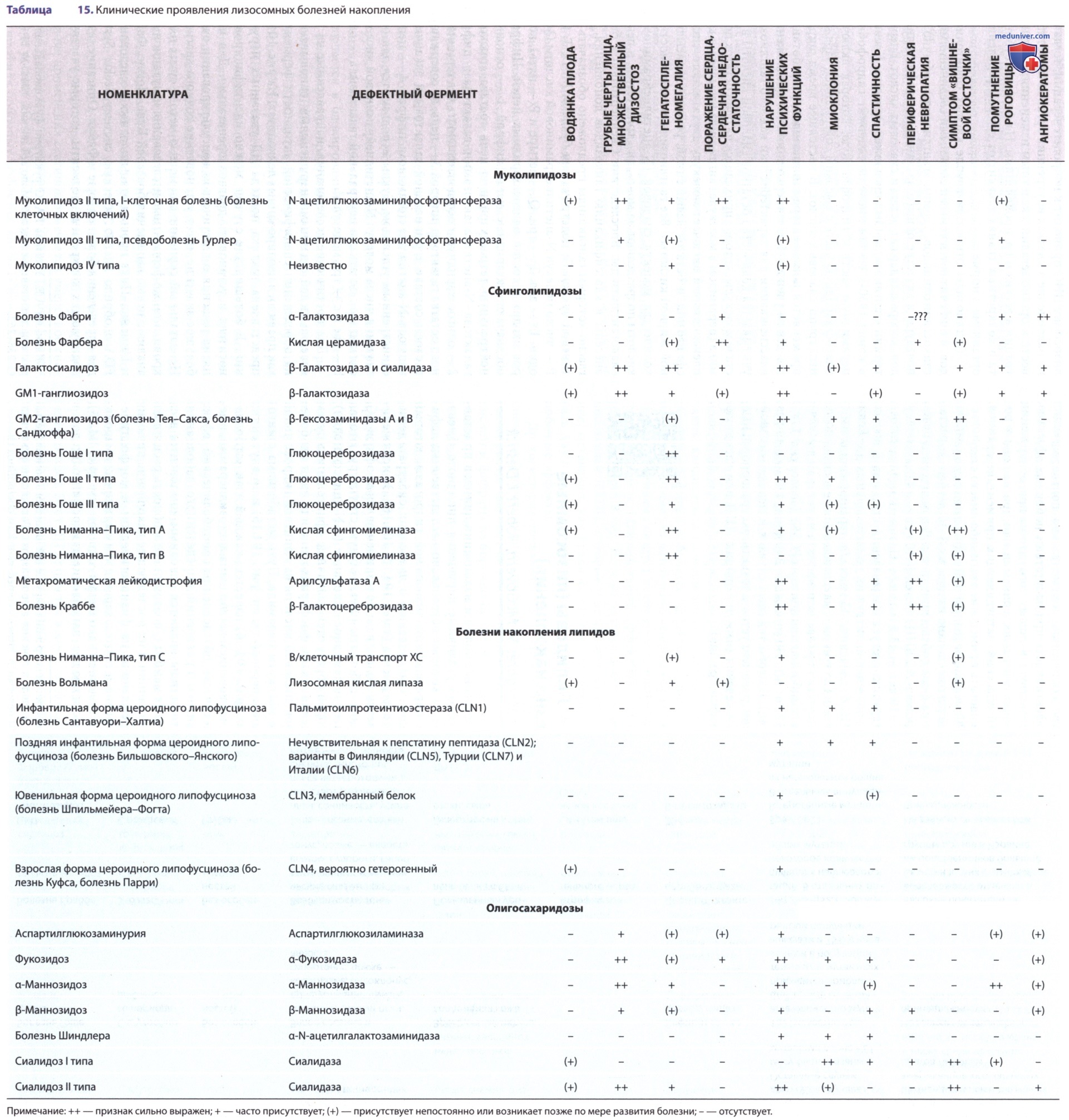
У пациентов с инфантильной формой болезни Тея-Сакса в грудном возрасте развиваются такие клинические проявления, как потеря двигательных навыков, усиление четверохолмного рефлекса (старт-рефлекс, проявляется вздрагиванием на неожиданный раздражитель), бледность макулы и симптом «вишневой косточки» в области сетчатки (см. табл. 15). Как правило, у детей с этим заболеванием развитие соответствует возрасту до 4-5 мес, когда у них замечают ослабление зрительного контакта и чрезмерную реакцию на шум (гиперакузию).
Может развиваться макроцефалия, не связанная с гидроцефалией. На втором году жизни развиваются судорожные припадки, которые могут быть рефрактерны к терапии антиконвульсантами. Процесс нейродегенерации не прекращается, и летальный исход наступает в возрасте 4 или 5 лет. Также имеются данные о более раннем развитии судорожного синдрома, к 8-10 мес, отмечаются генерализованные тонико-клонические, парциальные приступы, резистентные к терапии с последующим прогрессированием поражения ЦНС и развитием псевдобульбарного синдрома к 2 годам. Ювенильная форма и форма с более поздним началом в первое время проявляются атаксией и дизартрией. При этом может не наблюдаться симптома «вишневой косточки».
Поздняя инфантильная (ювенильная) форма развивается в возрасте от 2 до 10 лет, первоначально появляются нарушение походки, неустойчивость в позе Ромберга с последующим развитием тремора конечностей и атактического синдрома. По мере постепенного прогрессирования с возрастом развивается дизартрия, судорожный синдром, миоклонии, утрата интеллектуальных и моторных навыков. Продолжительность жизни составляет в среднем 10-15 лет от начала проявлений болезни. При взрослой форме болезни Тея-Сакса клинические проявления выявляются на 1-2-м десятилетии жизни. Характерными признаками считаются атактический синдром, дизартрия, выраженные психиатрические нарушения (возможны тяжелые психозы).
Клинические проявления болезни Сандхоффа сходны с проявлениями болезни Тея-Сакса. Выделяют три формы болезни Сандхоффа (Зандхоффа): инфантильную (самую частую), подострую и хроническую. При инфантильной форме клинические проявления развиваются на первом году, в первые месяцы жизни.
В грудном возрасте у детей с болезнью Сандхоффа отмечаются гепатоспленомегалия, поражение сердца и незначительные костные аномалии.
Ювенильная форма этого заболевания проявляется атаксией, дизартрией и нарушением психических функций, однако при этом не наблюдают увеличения внутренних органов или симптома «вишневой косточки». Эта форма очень редко встречается, заболевание манифестирует в возрасте от 3 до 10 лет, формируется умственная отсталость, прогрессирующая спастичность, слепота за счет атрофии зрительного нерва и пигментной дегенерации сетчатки, на терминальной стадии течения формирование децеребрационной ригидности.
Хроническая форма также очень редка, возраст начала заболевания либо от 3 до 10 лет, либо на 1-2-м десятилетии. Клиническая картина вариабельна, возможна прогрессирующая дистония или спиноцеребеллярная дегенерация, болезнь моторных нейронов или психозы.
Лечения болезни Тея-Сакса или болезни Сандхоффа не существует, однако изучаются экспериментальные подходы.
Как правило, диагноз инфантильной формы болезни Тея-Сакса и болезни Сандхоффа предполагают при наличии у младенца соответствующих отклонений со стороны НС и симптома «вишневой косточки». Окончательный диагноз устанавливают путем определения активности β-гексозаминидазы А и В в лейкоцитах периферической крови. Эти два заболевания различают по результатам исследования ферментной активности.
При болезни Тея-Сакса отмечается только дефицит фермента β-гексозаминидазы А, а при болезни Сандхоффа — дефицит и β-гексозаминидазы А, и β-гексозаминидазы В.
Риск развития обоих заболеваний можно диагностировать пренатально путем определения уровней ферментов в клетках плода, полученных путем амниоцентеза или взятия образцов ворсин хориона. Выявить носителей заболевания в семьях также можно путем определения уровней β-гексозаминидаз А и В. Действительно, при болезни Тея-Сакса скрининг на носительство рекомендуется проводить до наступления беременности всем парам, в которых хотя бы один член семьи является евреем-ашкенази, чтобы выявить пары, подверженные риску.
Эти исследования можно провести методом определения активности β-гексозаминидазы А в лейкоцитах периферической крови или плазме. Кроме того, чтобы выявить конкретный молекулярный дефект, необходимо проводить молекулярные исследования у носителей, выявленных методом определения активности ферментов. Это позволит более точно идентифицировать носителей в семье и провести пренатальную диагностику в парах из группы риска с определением уровня ферментов и генотипа.
После введения скрининговых программ в популяции евреев-ашкенази показатели заболеваемости болезнью Тея-Сакса значительно снизились. У новорожденных скрининговые исследования можно проводить методом определения специфических гликосфинголипидных маркеров или активности соответствующих ферментов в сухих пятнах крови.