GM1-ганглиозидоз у ребенка - кратко с точки зрения педиатрии
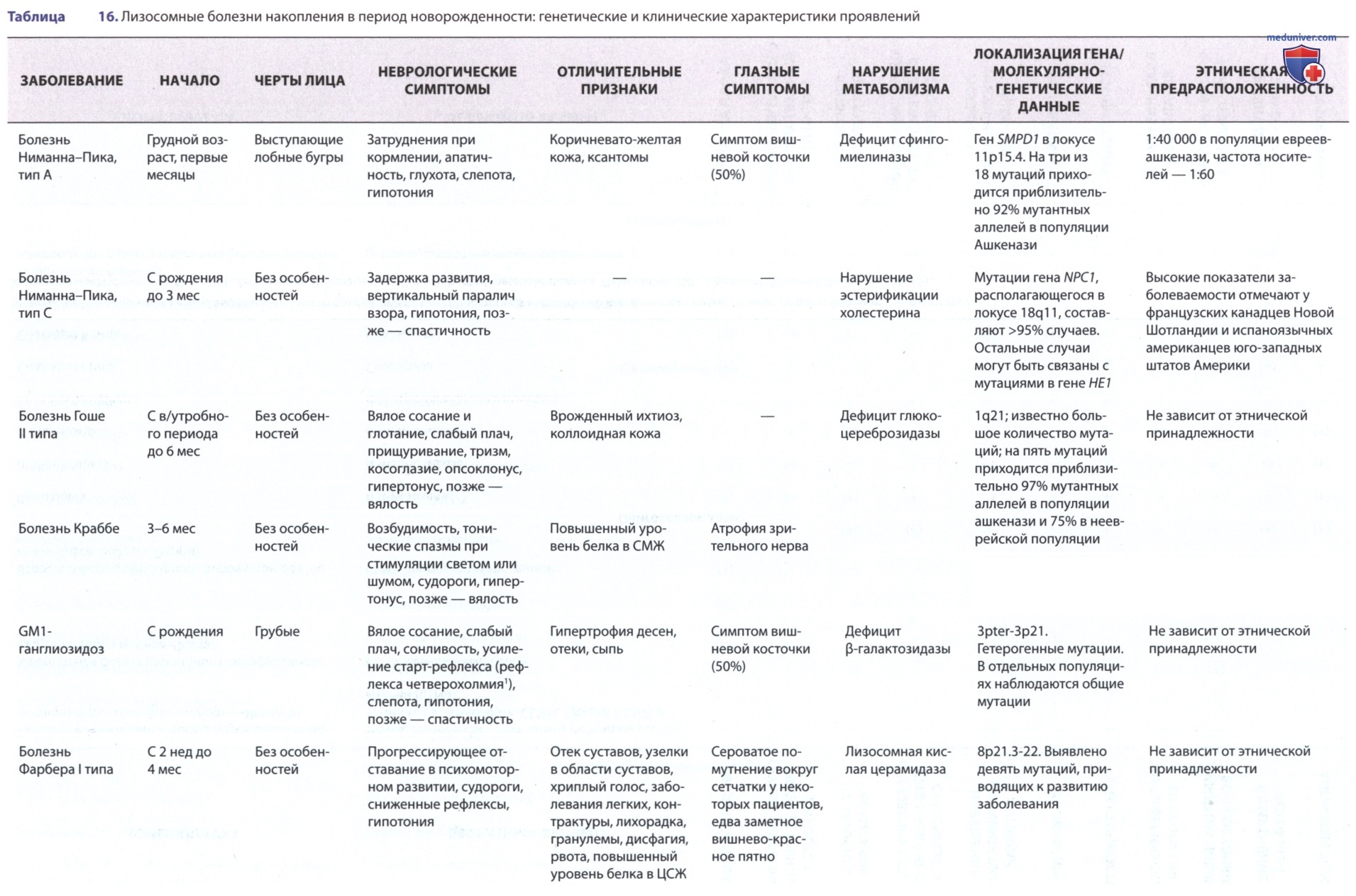
GM1-ганглиозидоз чаще всего проявляется в раннем детстве, но также описаны ювенильный подтип и подтип с началом во взрослом возрасте. Для заболевания характерен АуР-тип наследования. Каждый из подтипов возникает в результате различных генных мутаций, которые приводят к дефициту активности β-галактозидазы — лизосомного фермента, который кодируется геном, расположенным в третьей хромосоме (3p21.33).
Хотя для данного нарушения характерно патологическое накопление GM1-ганглиозидов как в лизосомах нервных клеток, так и в лизосомах клеток внутренних органов, оно наиболее выражено в клетках ГМ. Кроме того, у пациентов с GM1-ганглиозидозом накапливается в печени и выводится с мочой кератинсульфат, представляющий собой мукополисахарид.
Ген β-галактозидазы был выделен и секвенирован; мутации, вызывающие подтипы заболевания, также идентифицированы. В настоящее время выделяют основную по распространенности острую инфантильную форму, ювенильную и взрослую*.
P.S. * Захарова Е.Ю., Байдакова Б.Г., Михайлова С.В. и др. Лизосомные болезни накопления: Руководство для врачей. М.: ГЭОТАР-Медиа, 2021.
У новорожденных можно обнаружить такие клинические проявления инфантильной формы GM1-ганглиозидоза, как гепатоспленомегалия, отеки и кожная сыпь (ангиокератома). Чаще всего заболевание проявляется в первые 6 мес жизни задержкой развития с последующим прогрессированием отставания в психомоторном развитии и появлением тонико-клонических судорог.
Типичное лицо характеризуется низкой посадкой ушных раковин, выступающими лобными буграми, вдавленной переносицей и аномально длинным фильтром. Почти у 50% пациентов имеется симптом «вишневой косточки» в макулярной зоне. Отмечаются гепатоспленомегалия и аномалии скелета, сходные с изменениями при мукополисахаридозах, в т.ч. клювовидные выступы в передних отделах позвонков, расширение турецкого седла и утолщение костей свода черепа. К концу первого года жизни большинство пациентов становятся слепыми и глухими.
У них развиваются тяжелые неврологические нарушения с децеребрационной ригидностью. Смерть, как правило, наступает в возрасте 3-4 лет.
Выделены главные ДДК инфантильной формы:
• гепатоспленомегалия;
• дегенерация макулы, симптом «вишневой косточки»;
• изменения лица по типу «гаргоилизма»;
• утолщение кожи, отек кожи;
• множественный дизостоз;
• прорессирующие психомоторные расстройства;
• гиперэкскреция олигосахаридов;
• недостаточность β-галактозидазы в лейкоцитах и фибробластах*.
P.S. * Захарова Е.Ю., Байдакова Б.Г., Михайлова С.В. и др. Лизосомные болезни накопления: Руководство для врачей. М.: ГЭОТАР-Медиа, 2021.
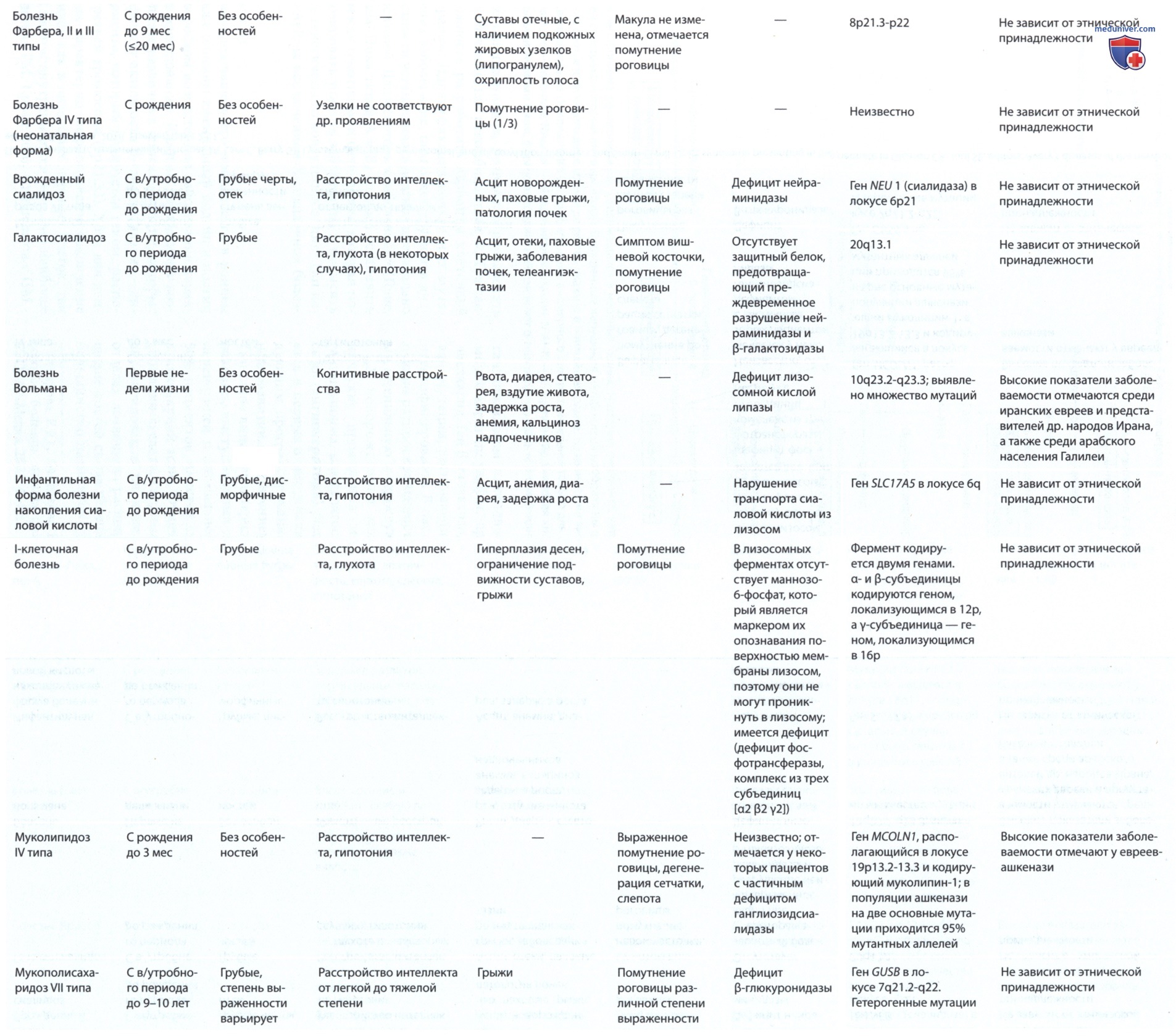
При ювенильной форме GM1-ганглиозидоза клиническая картина отличается. Заболевание может начинаться в разном возрасте. При этой форме ганглиозидоза возраст дебюта варьирует от грудного возраста до 2-6 лет. У пациентов отмечаются в основном неврологические симптомы, в т.ч. атаксия, дизартрия, нарушение интеллекта и спастичность. Заболевание прогрессирует медленно. Пациенты могут пережить четвертую декаду жизни. У таких пациентов не поражаются внутренние органы, отсутствуют аномалии лица и характерные изменения со стороны скелета, которые наблюдают при первом типе заболевания.
Главными ДДК ювенильной формы GM1-ганглиозидоза считаются:
• динамическая атаксия, нарушение походки;
• дизартрия, афазия, судороги;
• изменения лица по типу «гаргоилизма»;
• прогрессирующие психомоторные расстройства;
• недостаточность β-галактозидазы в лейкоцитах и фибробластах.
При взрослой (хронической) форме GM1-ганглиозидоза заболевание манифестирует в первые 10 лет жизни (от 3 до 8 лет) мозжечковой симптоматикой (динамическая атаксия, дизартрия). Далее присоединяется пирамидная и экстрапирамидная симптоматика, развиваются торсионно-дистонические гиперкинезы, атипичная спинальная мышечная атрофия или атипичная спиноцеребеллярная дегенерация, возможны «церебральные параличи» с прогрессирующей гипотонией мышц конечностей.
Заболевание часто трактуется как «ювенильный паркинсонизм», судороги бывают редко, острота зрения не снижается. Описаны пациенты со взрослой формой заболевания, у которых имелись нарушения походки и речи, дистония и легкие отклонения со стороны скелета.
Главными ДДК взрослой формы GM1-ганглиозидоза считаются:
• торсионно-дистонические гиперкинезы;
• спиноцеребеллярная дегенерация;
• «церебральный паралич» (подобие детского церебрального паралича);
• недостаточность β-галактозидазы в лейкоцитах и фибробластах.
Ни для одной из форм GM1-ганглиозидоза не существует специфического лечения.
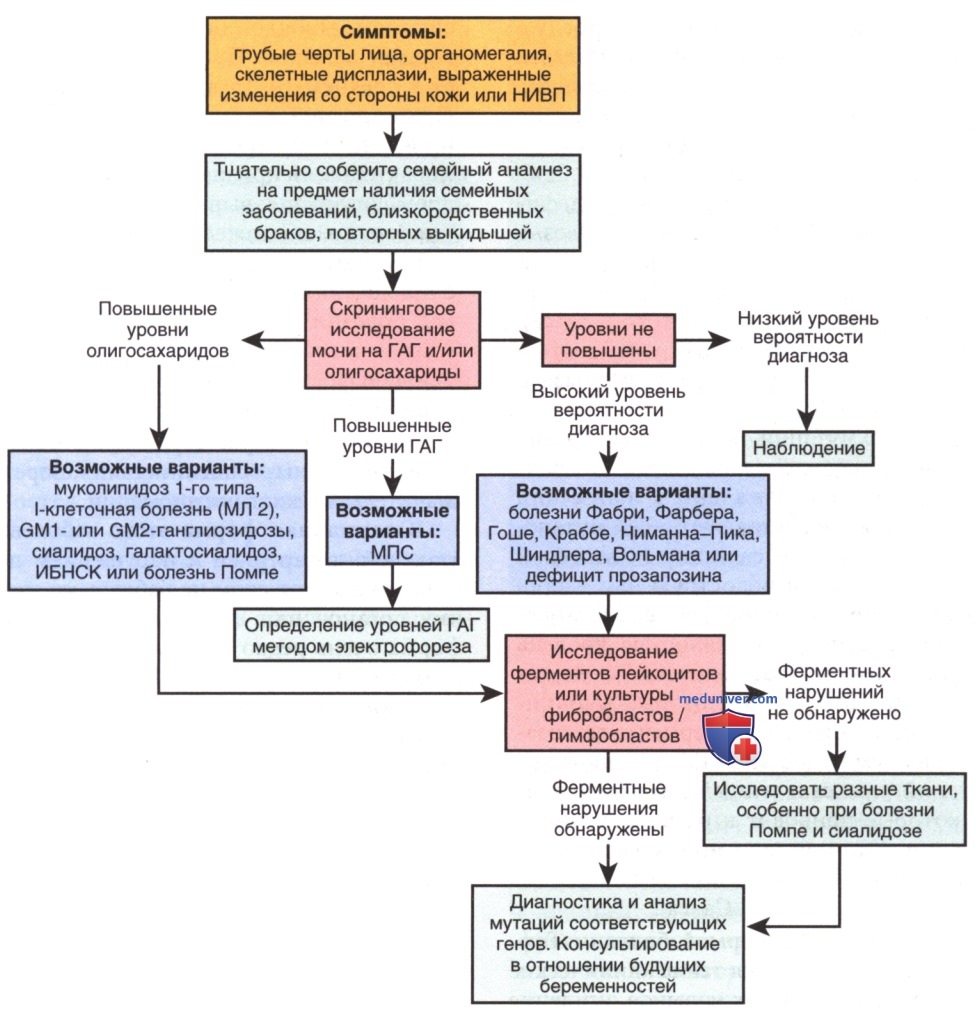
Рисунок 17. Алгоритм клинического обследования детей грудного возраста с подозрением на лизосомные болезни накопления. ГАГ — гликозаминогликаны; ИБНСК — инфантильная форма болезни накопления сиаловой кислоты; НИВП — неиммунная водянка плода
Диагноз GM1-ганглиозидоза следует предполагать у детей грудного возраста с типичными клиническими признаками, а для его подтверждения необходимо выявить дефицит активности β-галактозидазы в лейкоцитах периферической крови. Также при лабораторном исследовании можно выявить вакуолизацию лимфоцитов, «пенистые клетки» в биоптате печени, костного мозга и пр. Др. заболевания, которые имеют некоторые общие черты с ганглиозидозами ГМ 1, включают болезнь Херлера (мукополисахаридоз типа I), болезнь 1-клеток и болезнь Ниманна-Пика типа А.
Каждое из этих заболеваний можно отличить по критерию выявления специфического ферментативного дефицита. Носительство заболевания диагностируют методом определения ферментной активности в лейкоцитах периферической крови или путем идентификации специфических генных мутаций. Пренатальную диагностику проводят методом определения активности ферментов в культуре амниоцитов или ворсинок хориона или же методом выявления мутаций, вызывающих соответствующее заболевание.
Пациентам с GM1-ганглиозидозом проводят только поддерживающую терапию. Однако в исследованиях на мышах с GM1--ганглиозидозом продемонстрировано, что N-октил-4-эпи-β-валиенамин, который при пероральном применении стабилизирует белок мутантного фермента, продуцируемого пораженными животными, пересекает ГЭБ и замедляет прогрессирование неврологических симптомов. Это свидетельствует о том, что научные исследования при участии человека в данной области м.б. потенциально эффективными.