Болезнь Гоше представляет собой мультисистемный липидоз. Это заболевание характеризуется отклонениями гематологических показателей, органомегалией и вовлечением скелета. Последние, как правило, проявляются болями в костях и патологическими переломами (см. табл. 15).
Болезнь Гоше — одна из самых распространенных лизосомных болезней накопления и является наиболее частым наследственным заболеванием в популяции евреев-ашкенази.
Она считается наиболее частой формой лизосомных болезней накопления, обусловленной дефектом гена GBA, кодирующего лизосомный фермент β-D-глюкозидазу (глюкоцереброзидазу), ответственную за катаболизм липидов. Эта наследственная патология считается панэтнической, ее частота варьирует от 1:40 000 до 1:70 000. В популяции последних (выходцев из Восточной Европы) ее распространенность (особенно 1-го типа) наиболее высокая и составляет 1:450-1:2500 новорожденных.
Существует три клинических подтипа, которые различают по отсутствию или наличию и прогрессированию неврологических проявлений: тип 1, или взрослая, ненейронопатическая форма; тип 2, или острая нейронопатическая форма; и тип 3, ювенильная, или подострая или нейронопатическая форма. Тип наследования при всех трех формах АуР. Тип 1, на который приходится 99% случаев, характеризуется поразительной предрасположенностью евреев-ашкенази. Показатели заболевания в этой популяции составляют ~1:1000 живорожденных, а частота встречаемости носителей - ~1:18 взрослых.
В нееврейской популяционной группе частота гетерозиготного носительства мутантного аллеля составляет от 1:640 до 1:3969, причем в некоторых странах преобладают разные клинические подтипы болезни Гоше. В Европе, Канаде, США и Австралии чаще встречается болезнь Гоше 1-го типа, а в Египте (палестинские арабы), Японии, Северной Швеции (популяция Норрботтен) и Польше выше заболеваемость разл. вариантами хронической нейронопатической формы с частотой приблизительно 1:50 000.
Болезнь Гоше развивается в результате дефицита активности лизосомной гидролазы, кислой β-гликозидазы, которая кодируется геном, локализованным на хромосоме 1q21-q31. Дефект фермента приводит к накоплению нерасщепленных гликолипидных субстратов, в частности гликозилцерамида в клетках РЭС. Возрастающее накопление указанных соединений приводит к инфильтрации костного мозга, прогрессирующей гепатоспленомегалии и вторичным изменениям костной системы.
Дефицит фермента кислой β-гликозидазы приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше). В результате имеющихся метаболических нарушений происходит хроническая активация макрофагальной системы, аутокринная стимуляция моноцитопоэза и увеличение абсолютного количества макрофагов с нарушением регуляторных функций макрофагов.
У пациентов, принадлежащих к популяции евреев-ашкенази, на четыре мутации — N370S, L444P, 84insG и IVS2+2 — приходится приблизительно 95% мутантных аллелей. Это позволяет проводить скрининг на болезнь Гоше в данной популяции. Наблюдаемые корреляции между генотипом и фенотипом служат молекулярной основой клинической гетерогенности при болезни Гоше 1-го типа.
Среди гомозиготных пациентов по мутации N370S отмечают тенденцию к более позднему началу клинических проявлений и медленному прогрессированию заболевания, чем у пациентов с одной копией N370S и др. распространенным мутантным аллелем.
В настоящее время описано более 450 разл. мутаций.
Клинические проявления болезни Гоше 1-го типа характеризуются разным возрастом начала — от раннего детского до пожилого возраста. При этом симптомы заболевания наблюдают в основном у подростков. На момент обращения у пациентов могут быть экхимозы, обусловленные тромбоцитопенией, хроническая усталость, связанная с анемией, гепатомегалия с повышенными показателями функциональных печеночных проб, спленомегалия и боли в костях. В некоторых случаях у пациентов на момент обращения отмечают поражение легких. Пациенты, у которых заболевание манифестирует в первой декаде жизни, часто не относятся к еврейской популяции.
У них отмечают задержку роста и более злокачественное течение. У др. пациентов диагноз может быть установлен случайно, при проведении обследования по поводу др. заболеваний или при проведении рутинных исследований. У таких пациентов может быть более легкое или даже доброкачественное течение. У пациентов с клиническими проявлениями спленомегалия прогрессирует и может достигать значительных размеров. У большинства пациентов развиваются рентгенологические признаки вовлечения костной системы, в т.ч. колбообразная деформация дистальных отделов бедренных костей и проксимальных отделов большеберцовых костей (колбы Эрленмейера).
Клинические признаки вовлечения костей, которые наблюдаются у большинства пациентов, представлены болями в костях, псевдоостеомиелитом или патологическими переломами. В длинных костях, в т.ч. в бедренной кости, ребрах и костях таза, могут развиваться очаги остеолизиса. В раннем возрасте могут обнаруживаться признаки остеосклероза. Могут возникать костные кризы с выраженными болями и отеком. На фоне тромбоцитопении могут возникать носовые кровотечения или кровоизлияния. На них часто не обращают внимания, пока не появляются др. симптомы. Развитие и интеллект не страдают, за исключением детей с выраженной задержкой роста и развития на фоне хронического заболевания.
Болезнь Гоше 1-го типа ранее считали взрослым клиническим вариантом, но в большинстве случаев (56%) дебют болезни развивался в первое и второе десятилетие жизни, у 68% пациентов и у половины (48%) имел место до 6 лет. У взрослых больных в 28% случаев заболевание начиналось в возрасте 30-40 лет и в 50-80 лет в 17% случаев.
Наиболее распространенные симптомы болезни Гоше 1-го типа: спленомегалия (95%), гепатомегалия (87%), рентгенологически выявленные костные изменения (81%), тромбоцитопения (50%), анемия (40%), задержка роста (34%), хронические боли в костях (27%) и костные кризы (9%). Отличием болезни Гоше 1-го типа от нейронопатических форм является отсутствие раннего специфического поражения ЦНС, хотя периферическая невропатия и синдром паркинсонизма выявлены в ряде случаев у больных с этим типом заболевания.
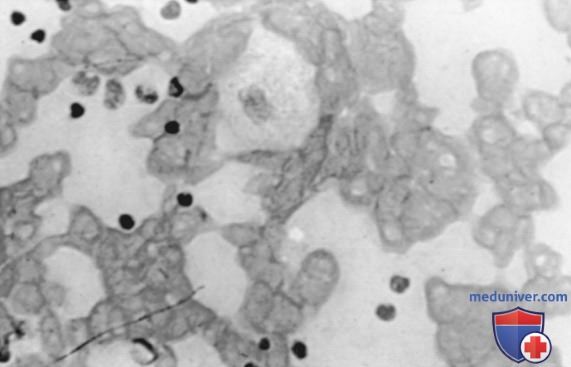
Патогист. маркером болезни Гоше являются клетки Гоше в РЭС, в частности в костном мозге (рис. ниже).
Клетки селезенки пациента с болезнью Гоше. Показаны характерные клетки селезенки, нагруженные глюкоцереброзидом
Глюкоцереброзидаза содержится во всех клетках организма, но макрофаги наиболее чувствительны, т.к. одной из основных их функций является деградация клеток крови, закончивших жизненный цикл. Низкая активность или отсутствие β-глюкоцереброзидазы способствуют накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления — клеток Гоше. Эти клетки, диаметр которых составляет 20-100 мкм, по внешнему виду напоминают скомканную бумагу, цитоплазма имеет типичный «сморщенный» или полосатый вид. Это связано с накоплением субстрата в цитоплазме. При окрашивании периодической кислотой-реактивом Шиффа (ПКШ) отмечается выраженная положительная реакция цитоплазмы клеток Гоше.
При наличии этих клеток в костном мозге и образцах ткани вероятность болезни Гоше очень высока, хотя эти клетки можно обнаружить и у пациентов с гранулоцитарным лейкозом и миеломой.
Болезнь Гоше 2-го типа является редкой клинической формой, заболеваемость не зависит от этнической принадлежности. Это заболевание характеризуется быстрым развитием нейродегенеративных изменений, выраженным вовлечением внутренних органов и развитием летального исхода в раннем возрасте. В грудном возрасте проявления болезни Гоше 2-го типа включают повышение тонуса, косоглазие и органомегалию. Типичны задержка роста и стридор, связанный с ларингоспазмом. Летальный исход, как правило, наступает на фоне дыхательной недостаточности после нескольких лет прогрессирующей задержки психомоторного развития.
Известны два клинических варианта болезни Гоше 2-го типа. При перинатально-летальном варианте (форме) во время беременности матери развивается в/утробная водянка плода с высокой вероятностью антенатального летального исхода или преждевременными родами с гибелью новорожденного в ближайшие сутки после рождения. При срочных родах ребенок погибает в течение первого квартала в связи с развитием РДС. Частым симптомом этого варианта болезни Гоше 2-го типа является ихтиоз, наиболее выраженный на ступнях, ладонях и в местах кожных складок. В клинический фенотип входят неврологические и висцеральные нарушения, дис-морфологические изменения лица.
Второй клинический вариант болезни Гоше 2-го типа представляет собой раннюю младенческую форму, которая дебютирует в первые 6 мес жизни симптомокомплексом, который включает следующие признаки поражения внутренних органов и ЦНС: выраженную гепатоспленомегалию; бульбарный синдром (дисфонию, дисфагию, дизартрию); тризм; билатеральное фиксированное косоглазие; прогрессирующие пирамидные нарушения (тетрапарез, гиперрефлексию, спастичность мышц с ретракцией шеи, положительный симптом Бабинского и др. патологические рефлексы); прогрессирующую задержку психомоторного развития и утрату ранее приобретенных навыков; тонико-клонические и др. типы судорожных приступов, резистентных к терапии антиконвульсантами.
Наиболее тяжелой формой течения болезни Гоше 2-го типа считается перинатально-летальный вариант, проявления которого существенно отличаются от классических симптомов 2-го типа. Известно ~50 описанных случаев этого варианта. По данным молекулярно-генетических исследований, данная форма ассоциируется с нулевыми аллелями, которые приводят к значительному снижению ферментативной активности, и частый аллель L444P присутствует в виде рекомбинантного аллеля (напр., RecNcil (p.L483P-p.A495P-p.V499V). Прогноз при болезни Гоше 2-го типа неблагоприятный, летальный исход связан с ДН на фоне аспирационной пневмонии или РДС при наличии бульбарных неврологических расстройств (нарушение глотания, поперхивание) и варьирует от раннего неонатального периода до 4 лет (в среднем 9 мес).
Клинические проявления болезни Гоше 3-го типа занимают промежуточное положение между проявлениями при болезни Гоше 1-го и 2-го типа. Начало болезни варьирует до 12-13 лет, а летальный исход может наступить в возрасте 10-15 лет. Дебют может варьировать от 1 мес до 14 лет. Высокие показатели заболеваемости до 1 на 50 000 отмечаются среди представителей населения провинции Норботтен в Швеции. Характерно поражение НС.
При болезни Гоше 3-го типа выраженность гепатоспленомегалии выше, чем при первом типе этого заболевания, поражение осевого скелета включает бочкообразную деформацию ГК и кифосколиоз позвоночника. Также при болезни Гоше 3-го типа неврологическая симптоматика развивается отсроченно, после висцеральных, костных и гематологических проявлений заболевания. Изменения со стороны НС вариабельны, основными и ранними неврологическими проявлениями болезни Гоше 3-го типа являются глазодвигательные расстройства — окуломоторная апраксия и/или косоглазие, которые длительно могут быть моносимптомами болезни; миоклонии, постепенно нарастающими и переходящими в генерализованные тонико-клонические судороги.
Далее прогрессирует экстрапирамидная ригидность с нарастающим снижением интеллектуальных функций вплоть до деменции, развиваются расстройства письма и речи, диффузная мышечная гипотония трансформируется в спастичность. Возможны поведенческие расстройства, эпизоды психоза1.
Болезнь 3-го типа далее подразделяют на типы 3а и 3b на основании степени вовлечения НС и наличия прогрессирующей миотонии и деменции (тип 3а) или изолированного надъядерного паралича взора (тип 3b).
В КР РФ выделяют три варианта болезни Гоше 3-го типа. 3А-тип отличается преобладанием неврологических проявлений и обычно развивается до 10 лет или в подростковом возрасте. Имеется описание клинического случая больной 17 лет, представленного Botross N.P. и соавт., с характерными генерализованными миоклоническими приступами, атаксией, окуло-моторной апраксией, когнитивными расстройствами и отсутствием гепатоспленомегалии и костных повреждений. У больных при 3B-типе болезни Гоше преобладают поражение внутренних органов и костно-суставной системы, а симптомы со стороны ЦНС проявляются только глазодвигательными расстройствами в виде окуло-моторной апраксии и страбизма.
Типичным для 3B-типа считается формирование в раннем детстве гепатоспленомегалии, признаки со стороны костной системы включают деформации ГК и выраженный кифосколиоз без костных болей и костных кризов.
3С-тип болезни Гоше (сердечно-сосудистая форма) считается наиболее редким вариантом хронической нейронопатической формы, для которого характерны гепатоспленомегалия, неатеросклеротическое поражение сердца и крупных сосудов в виде кальцификации сердечных клапанов, аорты и КА и развитие ЗСН, окуломоторная апраксия, помутнение роговицы; задержка умственного развития, гидроцефалия и эпилепсия, рефрактерные к стандартной терапии антиконвульсантами. Продолжительность жизни при ЗС-типе болезни Гоше определяется клиническим вариантом (типом) и варьирует от 4 до 60 лет.
Главная особенность болезни Гоше 3-го типа заключается в сочетании с поражением паренхиматозных органов (гепатоспленомегалия) неврологической симптоматики, сходной с проявлениями болезни Гоше 2-го типа, но менее выраженной и развивающейся в возрасте от 6 до 15 лет и позже.
Также выделен промежуточный тип болезни Гоше (между 2-м и 3-м типами) с тяжелыми неврологическими проявлениями в более позднем возрасте и поражением внутренних органов. Возраст дебюта при данном типе болезни Гоше варьирует от года до 7 лет. Goker-Alpan О. и соавт. (2003 г.) описано 9 случаев детей с промежуточным типом с манифестацией симптомов в возрасте 1,5 года. Эпилептические приступы у пациентов с этом клиническим типом резистентны к противосудорожной терапии. Заболевание быстро прогрессирует и приводит к смерти больных в возрасте 2-7 лет в связи с поражением ствола ГМ и аспирационной пневмонией.
Редкие проявления болезни Гоше включают поражение легких, развитие гошером (псевдоопухолей) и моноклональной гаммапатии. Поражение легких с развитием легочной гипертензии имеет место в 1% случаев в основном у больных после спленэктомии. Патогенез поражения легких предположительно обусловлен инфильтрацией макрофагами сосудов легких и альвеол. Гошеромы представляют собой опухолеподобные образования, состоящие из скоплений клеток Гоше или заместившего их фиброза, хорошо визуализируемые при УЗИ и КТ с контрастированием. Гошеромы являются доброкачественными и требуют динамического наблюдения.
Риск формирования моноклональной гаммапатии с развитием миеломной болезни статистически выше при болезни Гоше, но это относится к взрослой категории больных.
В настоящее время классификация болезни Гоше пересматривается в связи с описанием разл. клинических случаев. Наиболее актуальна сейчас концепция фенотипического континуума.
При болезни Гоше выявлены определенные генофенотипические корреляции. Установлено, что мутация c.1226A>G, p.Asn409Ser (p.Asn370Ser по старой номенклатуре) в гомозиготном состоянии или в комбинации с любым др. аллелем приводит к болезни Гоше l-го типа, а инактивирующие точечные мутации, рекомбинантные аллели и крупные делеции ассоциируются с нейронопатическими формами заболевания. Мутация с.1448Т>С,р. Leu483Pro (p.Leu444Pro по старой номенклатуре) является одной из наиболее частых и описана при болезни Гоше 2-го типа.
а) Клиническая диагностика. При сборе анамнеза и жалоб следует обратить внимание на наличие:
• семейного анамнеза (наличие спленэктомии или перечисленных выше симптомов у родных братьев и сестер);
• задержки физического и полового развития;
• слабости, повышенной утомляемости, частых респираторных инфекций;
• проявлений спонтанного геморрагического синдрома (в виде п/к гематом, кровоточивости слизистых оболочек) или длительных кровотечений при малых оперативных вмешательствах;
• болей в костях и суставах (давность, характер и локализацию болей, наличие в прошлом переломов костей);
• предшествующей спленэктомии (полной или частичной);
• неврологической симптоматики (глазодвигательная апраксия или сходящееся косоглазие, атаксия, потеря интеллекта, нарушения чувствительности и др.).
б) Физикальное обследование. Пациенту с подозрением на болезнь Гоше следует провести всестороннее физикальное обследование, обращая особое внимание на пальпаторное и перкуторное исследование печени и селезенки.
Для болезни Гоше 2-го и 3-го типов характерны следующие признаки:
• задержка физического развития;
• задержка психомоторного развития;
• геморрагический синдром;
• сплено-/гепатомегалия;
• асцит;
• косоглазие;
• судороги;
• костные боли (костные кризы);
• нарушение подвижности в суставах, обусловленное асептическим некрозом;
• патологические переломы;
• астенический синдром;
• снижение интеллекта (от незначительных изменений до тяжелой деменции);
• экстрапирамидная ригидность;
• мозжечковые нарушения;
• расстройства речи, письма;
• поведенческие изменения, эпизоды психоза;
• миоклонии, постепенно нарастающие и переходящие в генерализованные тонико-клонические судороги.
Для болезни Гоше 1-го типа характерны следующие признаки:
• задержка роста;
• сплено-/гепатоспленомегалия;
• геморрагический синдром;
• костные боли (костные кризы);
• нарушение подвижности в суставах, обусловленное асептическим некрозом;
• патологические переломы;
• задержка физического и полового развития;
• астенический синдром.
в) Лабораторная диагностика. У большинства больных с болезнью Гоше выявляются тромбоцитопения, лейкопения и анемия как проявления гиперспленизма. Поэтому всем больным, у которых предполагают наличие болезни Гоше, рекомендуется проведение развернутого ОАК (Hb, количество эритроцитов, цветовой показатель, количество лейкоцитов, тромбоцитов, лейкоцитарная формула и СОЭ).
При предположительном диагнозе болезнь Гоше рекомендуется проведение биохимического анализа сыворотки крови с определением АЛТ, ACT, общего билирубина ЩФ, ХС, ЛПВП, ЛПНП, железа, уровня ферритина, концентрации витамина В12, фолиевой кислоты, исследование уровня ионизированного кальция с целью подтверждения данного заболевания и проведения ДД с др. наследственными болезнями обмена в-в. При болезни Гоше в биохим. анализе сыворотки крови выявляются снижение концентрации липопротеидов, железа, витамина В12, фолиевой кислоты и ионизированного кальция.
При отсутствии лечения у больных с болезнью Гоше часто (87% случаев) отмечается гиперферритинемия на фоне нормального значения ОЖСС. Уровни ферритина коррелируют с тяжестью заболевания и наличием спленэктомии и значимо уменьшаются на фоне терапии. АЛТ, ACT и ЩФ повышены при болезни Гоше умеренно, отмечается преобладание ACT над АЛТ, причем корреляции с тяжестью поражения печени и степенью гепатомегалии не прослеживается.
Пациентам с клинической картиной болезни Гоше рекомендуется проведение коагулограммы с определением ПТВ в крови или в плазме, АЧТВ и МНО для оценки функционального состояния печени и свертывающей системы крови. При болезни Гоше в коагулограмме регистрируют удлинение АЧТВ и ПТВ. При болезни Гоше морфологическое исследование костного мозга не рекомендуется проводить детям в качестве первой линии диагностики.
Морфологическое исследование костного мозга способствует выявлению характерных диагностических элементов — клеток Гоше и позволяет исключить наличие гемобластоза или лимфопролиферативного заболевания как причины цитопении и гепатоспленомегалии. Но процедура забора клеток костного мозга является болезненной для пациента, а также результаты цитологического исследования м.б. неоднозначными. Имеется достаточно высокий риск л/о-данных костномозговой пункции при редком распределении клеток Гоше в миелограмме.
Л/о-результаты морфологического исследования миелограммы не позволяют полностью исключить болезнь Гоше, т.к. клетки Гоше чувствительны к механическим повреждениям и легко разрушаются в процессе приготовления и окраски мазков. Также важно то, что клетки-маркеры могут располагаться на периферии мазка костного мозга и не всегда попадают в поле зрения морфолога.
Возможны л/п-результаты исследования при обнаружении псевдоклеток Гоше (альтернативно активированных макрофагов), которые определяются при обширном перечне др. заболеваний. Перечень заболеваний, при которых в миелограмме м.б. псевдоклетки Гоше, включает множественную миелому, миелодисплазию, миело-диспластический синдром, хроническую миелоидную лейкемию, легочный туберкулез, микобактериозы, СКА, иммунную тромбоцитопению.
Всем пациентам с клиническими проявлениями болезни Гоше рекомендуется с целью верификации диагноза проведение биохимического исследования: определение активности β-D-глюкозидазы. Биохимическим критерием точного диагноза является снижение активности фермента β-D-глюкозидазы. В настоящее время диагностика болезни Гоше с определением активности фермента чаще проводится по пятнам сухой крови с использованием методов тандемной масс-спектрометрии.
Всем пациентам с клиническими проявлениями болезни Гоше и снижением активности фермента β-D-глюкозидазы с целью верификации диагноза рекомендовано проведение молекулярно-генетического исследования: выявление мутаций в гене GBA. Молекулярно-генетическая диагностика болезни Гоше позволяет обнаружить мутантные аллели гена GBA и состоит из двух этапов. На первом этапе происходит поиск наиболее распространенных мутаций с помощью скрининговых панелей (особенно в популяциях с преобладанием определенных мутаций), что позволит выявить до 80% значимых вариантов гена. На втором этапе проводят поиск редких и новых мутаций.
Всем пациентам с клиническими проявлениями болезни Гоше рекомендуется определение активности хитотриозидазы в плазме крови и уровня глюкозилсфингозина (Lyso-Gbl) для мониторинга терапии.
Дополнительные методы исследования при болезни Гоше включают определение концентрации и активности ряда биологически активных соединений, которые вырабатываются в избыточном количестве при активации макрофагов, нагруженных патологическим субстратом. На фоне терапии активность биомаркеров снижается за счет снижения накопленного глюкоцереброзида в сыворотке крови, клетках и тканях организма. Биомаркеры являются показателем эффективности терапии. Хитотриозидаза — наиболее чувствительный из известных биомаркеров, активность которого у больных болезнью Гоше повышена в десятки и даже сотни раз.
Однако активность данного фермента может быть повышена и при др. лизосомных болезнях накопления и воспалительных заболеваниях. Наряду с этим в 6-35% случаев в различных популяциях отмечается генетически обусловленный дефицит хитотриозидазы, поэтому данный фермент не всегда может быть использован для диагностики и мониторинга болезни Гоше. Хемокин CCL18/PARC также повышается при болезни Гоше, но не является специфичным маркером этого заболевания и может повышаться при онкопатологии, воспалительных процессах суставов, легких и кожи.
Новый биомаркер Lyso-Gbl обладает наибольшей специфичностью и чувствительностью и достоверно коррелирует с активностью хитотриозидазы и хемокина CCL 18/PARC в сочетании с гепатомегалией у больных с болезнью Гоше. XT и CCL18 — с гепатоспленомегалией у пациентов с болезнью Гоше. Значение Lyso-GLl более 420 нг/мл ассоциировано с тяжелым течением заболевания.
г) Инструментальные диагностические исследования. Всем пациентам с клиническими проявлениями болезни Гоше рекомендуется проведение рентгенографии трубчатых костей скелета для выявления и оценки тяжести поражения костно-суставной системы. Рентгенологическое исследование трубчатых костей проводится с целью диагностики деформаций скелета, переломов и определения толщины коркового слоя костей.
Изменения костной ткани трубчатых костей, характерные для болезни Гоше, могут включать истончение надкостницы, эндостальную зубчатость, пониженную трабекулярность, диффузный остеопороз, характерную колбообразную деформацию дистальных отделов бедренных и проксимальных отделов большеберцовых костей (колбы Эрленмейера), повышение рентгенопрозрачности костной ткани, наличие кистовидных просветлений, очаги остеолизиса, остеосклероза и остеонекроза и патологические переломы.
Рекомендовано проведение МРТ бедренных костей и ТБС в качестве надежного метода диагностики степени инфильтрации костного мозга и его структурных изменений при болезни Гоше при постановке данного диагноза, в динамике наблюдения и лечения для оценки эффективности последнего. МРТ позволяет выявлять признаки костного криза и очаги асептического некроза кости.
Остеоденситометрия поясничного отдела позвоночника рекомендуется больным с болезнью Гоше в качестве метода оценки выявления сниженной минеральной плотности костной ткани. Данный метод является «золотым стандартом» для поставленной цели, позволяет установить специфические изменения на ранних стадиях болезни Гоше и проводить мониторинг эффективности терапии. Степень снижения минеральной плотности костной ткани у детей определяется с помощью Z-критерия (Z-score) — количества стандартных отклонений (SD), на которое результат измерения отличается от средней величины для данного возраста и пола. Исследование проводится с 5-летнего возраста. Анализ результатов проводится на костный возраст ребенка, определяемый предварительно по рентгенограммам костей кистей.
При болезни Гоше рекомендовано проведение УЗИ и МРТ печени и селезенки с целью выявления их очаговых изменений и определения исходного объема органов для оценки течения патологического процесса и контроля эффективности заместительной ферментной терапии.
По данным литературы, у 20% пациентов в структуре органов выявляются крупные гипоэхогенные участки неправильной формы, однородной структуры, с нечетким размытым контуром и без четкой локализации — так называемые «узлы Гоше», или гошеромы.
При болезни Гоше рекомендовано проведение фиброэластометрии, которая представляет собой альтернативную неинвазивную ультразвуковую методику определения изменений паренхимы печени и селезенки. Этот метод представлен в клинических рекомендациях отдельных стран Европы для неинвазивной оценки структурных изменений печени при болезни Гоше. Также этот метод является достоверным индикатором состояния плотности органа, в данном случае изменений состояния паренхимы печени и прогрессирования фиброза, одного из главных показателей неблагоприятного исхода заболевания.
Рекомендовано проведение ЭКГ, допплер-ЭхоКГ, РОГК, определение функции внешнего дыхания у спленэктомированных пациентов с болезнью Гоше с высоким риском поражения легких для исключения легочной гипертензии и ДН. Также рекомендовано проведение эзофагогастродуоденоскопии при наличии соответствующих жалоб или признаков портальной гипертензии и проведение ЭЭГ у пациентов с неврологическими типами болезни Гоше.
В процессе обследования и комплексного лечения необходимо совместное ведение больного специалистами разных профилей. Пациентам с болезнью Гоше показаны первичные и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, травматолога-ортопеда, врача-гастроэнтеролога, врача-трансплантолога, врача-педиатра, врача-терапевта, а также врачей др. специальностей.
ДД с болезнью Гоше следует проводить у пациентов с органомегалией неуточненного генеза, легким появлением экхимозов, болями в костях или сочетанием указанных признаков. Клиническая гетерогенность проявлений болезни Гоше, широкая вариабельность возраста дебюта заболевания, неспецифичность ранних клинических проявлений диктуют необходимость проведения ДД с большим количеством гематологических, печеночных, костных, неврологических заболеваний и др. наследственными метаболическими болезнями.
Болезнь Гоше 1-го типа в зависимости от вида манифестации — разнообразные экзогенные и наследственные заболевания, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью.
Цитопенический и геморрагический синдромы чаще всего необходимо дифференцировать с лимфопролиферативными и миелопролиферативными заболеваниями, миелодиспластическим синдромом, анемиями др. этиологии, иммунной тромбоцитопений, тромбоцитопатиями и коагулопатиями (гемофилия). ДД гепатолиенального синдрома проводят с др. заболеваниями, характеризующимися увеличением печени и селезенки, прежде всего со сфинголипидозами (болезнь Ниманна-Пика типов A/В), заболеваниями печени с развитием цирроза и портальной гипертензии (в/утробные инфекции, вирусные гепатиты, аутоиммунный гепатит, болезнь Вильсона, гликогеновая болезнь, недостаточность альфа-1-антитрипсина, тирозинемия, дефицит лизосомной кислой липазы, гемохроматоз, гистиоцитоз, болезнь Фарбера).
При диагностике гепатоспленомегалии с неврологической симптоматикой (2-й и 3-й типы болезни Гоше) необходимо исключить все инфантильные формы сфин-голипидозов (болезнь Ниманна-Пика типа С), GM1-ганглиозидоз, галактосиалидоз, болезнь Вольмана, болезнь Фарбера (атипичные формы), а также врожденную окуломоторную апраксию1.
При исследовании костного мозга, как правило, обнаруживают клетки Гоше. Все предполагаемые диагнозы подтверждают путем определения активности кислой β-глюкозидазы в изолированных лейкоцитах или культуре фибробластов, а также путем выявления специфических мутаций в гене, кодирующем кислую β-глюкозидазу. Среди представителей популяции евреев-ашкенази носителей можно выявлять методом молекулярных исследований на общие мутации, которые часто встречаются в этой популяции. Всем членам семьи нужно предлагать пройти обследование. При этом следует учитывать, что гетерогенность даже среди членов одной семьи м.б. выражена настолько, что диагноз м.б. установлен даже у членов семьи, не имеющих симптомов.
Семьям с больными детьми рекомендуется медикогенетическое консультирование с целью разъяснения генетического риска. Как и при др. АуР-заболеваниях, при болезни Гоше для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики.
Пренатальная диагностика возможна путем определения активности фермента и/или специфических семейных мутаций в ворсинах хориона или культуре клеток амниотической жидкости.
Пренатальная диагностика проводится молекулярногенетическими или биохим. методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11-й неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22-й неделе беременности.
д) Лечение. Лечение пациентов с болезнью Гоше 1-го типа включает ферментную заместительную терапию (ФЗТ). Эффективность ФЗТ рекомбинантной человеческой кислой глюкозидазы с терминальными маннозными группами (имиглюцеразы, велаглюцеразы альфа или талиглюцеразы альфа) является стандартом оказания МП при лечении пациентов с болезнью 1-го типа. Основная часть симптомов (органомегалия, гематологические показатели, боли в костях) регрессирует на фоне ФЗТ (60 МЕ/кг) с в/в инфузиями, проводимыми 1 р/2 нед, костные проявления стабилизируются или уменьшаются. Хотя у пациентов с болезнью Гоше 2-го и 3-го типов ФЗТ не влияет на прогрессирование неврологических изменений, этот метод применяют в качестве паллиативного средства у отдельных пациентов, в частности у больных с болезнью Гоше 3-го типа с выраженными висцеральными проявлениями.
В КР РФ рекомендуется пожизненная ФЗТ рекомбинантной глюкоцереброзидазой пациентам с хроническим поражением НС при болезни Гоше 3-го типа, у которых имеются клинически значимые неневрологические проявления заболевания. При болезни Гоше 2-го типа не рекомендуется ФЗТ в связи с неэффективностью.
В РФ разработаны рекомендации по индивидуальному дозированию ЛП в зависимости от типа, клинического варианта и тяжести течения болезни Гоше, а также с учетом успешности достижения терапевтических целей на основании динамики клинических проявлений. Преимуществом ФЗТ является высокий профиль безопасности и эффективности для купирования основных проявлений болезни Гоше, к недостаткам относятся отсутствие влияния на неврологические симптомы (ЛП не проникают через ГЭБ) и частые инфузии, которые снижают качество жизни.
1. Принципы дозирования препаратов ферментной заместительной терапии. При болезни Гоше 1-го типа без поражения трубчатых костей скелета начальная доза имиглюцеразы составляет 30 ЕД/кг на 1 введение. При 2-м типе болезни Гоше с поражением трубчатых костей скелета (костные кризы, патологические переломы, очаги литической деструкции, асептический некроз головок бедренных костей) 60 ЕД/кг на 1 введение, а при 3-м типе болезни Гоше — 60 ЕД/кг на 1 введение.
Велаглюцераза альфа показана для длительного лечения детей с болезнью Гоше 1-го типа. Рекомендуемая доза составляет 30-60 ЕД/кг 1 р/2 нед. Доза корректируется индивидуально, в зависимости от достижения эффекта и его сохранения, однако применение доз выше 60 ЕД/кг не изучено.
Талиглюцераза альфа показана для лечения детей и взрослых с подтвержденным диагнозом болезни Гоше 1-го типа. Назначается детям в возрасте от 2 до 18 лет с висцеральными или гематологическими проявлениями. Рекомендуемая доза составляет 30-60 ЕД/кг 1 р/2 нед, в зависимости от клинической оценки, проведенной лечащим врачом.
При развитии проявлений остеопороза для замедления и прекращения потери костной массы, повышения ее прочности, предотвращения переломов костей в комплексной терапии рекомендуется применение ЛП витамина D [альфакальцидол, колекальциферол («Холекальциферол»), соли кальция] на фоне диеты, обогащенной кальцием.
Симптоматическая терапия при болезни Гоше 1-го типа включает также НПВС и антибактериальные ЛП для профилактики скелетных осложнений. В редких случаях при необратимых костных изменениях (остеоартрозы) показано проведение оперативного лечения: эндопротезирование суставов для купирования хронической боли и восстановления функции.
Не рекомендуется оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. При оперативных вмешательствах повышен риск кровотечения и инфицирования. Также не рекомендованы повторные пункции костного мозга и др. инвазивные диагностические мероприятия (биопсия печени, селезенки). Не рекомендовано проведение спленэктомии пациентам с болезнью Гоше. Противопоказано назначение ГКС с целью купирования цитопенического синдрома при доказанном диагнозе «болезнь Гоше».
Не рекомендовано и считается необоснованным назначение ЛП железа больным с развернутой картиной болезни Гоше, т.к. анемия в этих случаях носит характер «анемии воспаления»1.
Альтернативное лечение с помощью пероральных субстрат-редуцирующих ЛП, предназначенных для снижения синтеза глюкозилцерамида путем химического ингибирования синтазы глюкозилцерамида, включает миглустат, хотя его эффективность в отношении гематологических показателей не столь велика, как у ФЗТ. Однако эффективность этого ЛП в отношении гематологических показателей ниже, чем эффективность ФЗТ. Второй, более эффективный субстратный ингибитор, элиглустат, продемонстрировал значительную эффективность по сравнению с плацебо и не уступает имиглюцеразе, что делает его альтернативным пероральным препаратом первой линии для пациентов с болезнью Гоше 1-го типа. Небольшой группе пациентов была выполнена трансплантация костного мозга (ТКМ).
Эта операция обладает лечебным эффектом, но сопровождается большим процентом развития осложнений и высокими показателями летальности, что ограничивает выбор подходящих кандидатов.
2. Мониторинг состояния пациентов с болезнью Гоше. Ведение пациентов рекомендуется осуществлять в соответствии с рекомендациями по минимально необходимому мониторингу состояния больных при болезни Гоше I типа, разработанными Объединенной международной группой по изучению болезни Гоше (International Collaborative Gaucher Group). Контроль показателей крови необходимо проводить 1 раз в 3 мес, размеров паренхиматозных органов (УЗИ, MPT) — 1 раз в 6 мес, а также при изменении дозировки препарата или при значительных клинических осложнениях. Определение состояния костной ткани осуществляют 1 раз в год. Определение активности хитотриозидазы на фоне ферментной заместительной терапии проводят 1 раз в 12 мес.
Пациентов с болезнью Гоше рекомендуется наблюдать по месту жительства в амбулаторно-поликлинических условиях врачам-педиатрам, гематологам, с болезнью Гоше III типа — неврологам, при наличии костных нарушений — ортопедам до достижения возраста 18 лет.
Рекомендуется введение ФЗТ проводить регулярно при наличии показаний в случае осложненного течения болезни — в условиях круглосуточного стационара, в стабильном состоянии — в стационаре дневного пребывания или амбулаторно 1 р/2 нед. До достижения клинико-лабораторной ремиссии все пациенты с БГ должны проходить контрольное обследование с целью коррекции дозы ФЗТ в круглосуточном или дневном стационаре 2 раза в год; в дальнейшем обследование проводится 1 раз в год.