Болезнь Шиндлера у ребенка - кратко с точки зрения педиатрии
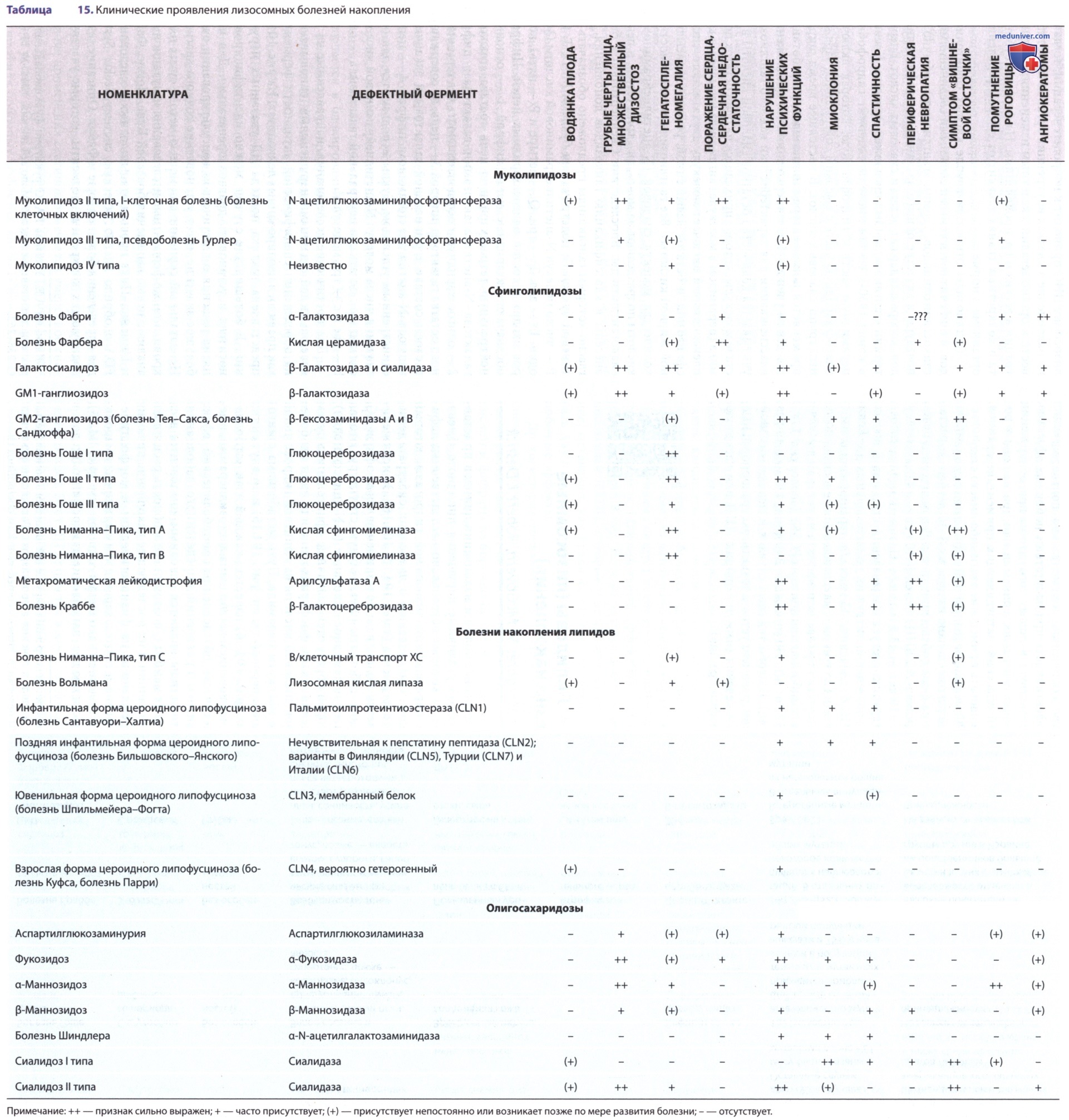
Болезнь Шиндлера представляет собой АуР-нейродегенеративное заболевание, обусловленное недостаточной активностью α-N-ацетилгалактозаминидазы и накоплением асиалогликопептидов и сиалилолигосахаридов (см. табл. 15). Ген, кодирующий фермент, располагается на хромосоме 22 (22q11).
Заболевание отличается клинической гетерогенностью, выделяют два основных клинических фенотипа. Болезнь типа I — нейроаксональная дистрофия с началом на 1-2-м году жизни.
Болезнь II типа характеризуется началом в разные возрастные периоды, легкими нарушениями интеллекта и наличием ангиокератом. Имеются данные о возрасте начала на третьем 10-летии жизни. Дебют заболевания проявляется развитием диффузной ангиокератомы, которая появляется в области нижних участков туловища с последующим распространением на все тело и лицо.
У больных имеются признаки «огрубения» черт лица — запавшая переносица, толстые губы, увеличенный кончик носа.
Дети с болезнью Шиндлера в первые 9-15 мес жизни развиваются нормально, а затем начинается быстро прогрессирующий нейродегенеративный процесс, приводящий к тяжелой задержке психомоторного развития, корковой слепоте и частым миоклоническим припадкам. Характерным симптомом считаются судороги. К концу второго года жизни развиваются косоглазие, атрофия зрительных нервов, нистагм, мышечная гипотония.
К возрасту 3-4 лет у больных имеет место тяжелая умственная отсталость, декортикационная поза и обездвиженность, сгибательные контрактуры всех суставов на фоне сохраняющихся миоклоний. По имеющимся данным, наибольший возраст продолжительности жизни — 8-9 лет*.
P.S. * Захарова Е.Ю., Байдакова Б.Г., Михайлова С.В. и др. Лизосомные болезни накопления: руководство для врачей. М.: ГЭОТАР-Медиа, 2021, 424 с.
Ни для одной из форм заболевания не существует специфического лечения. Диагноз устанавливают путем определения недостаточности фермента a-N-ацетилгалактозаминидазы в плазме, лейкоцитах или культуре кожных фибробластов или специфических генных мутаций.